
Lipid Therapy, CAC, and Sex Differences
Mechanistic and Clinical Differences Between PCSK9 Inhibitors, Statins, and Bempedoic Acid in Coronary Artery Calcium Progression, Plaque Biology, and Sex-Specific Cardiovascular Risk
The Evolution of the Atherosclerotic Paradigm
Historically, atherosclerosis was conceptualized as a simplistic, linear process of cholesterol accumulation within the arterial wall leading to mechanical obstruction of blood flow. Contemporary research has fundamentally shifted this understanding toward an immunoinflammatory paradigm. Atherosclerosis is now characterized as a chronic inflammatory disease, initiated by the subendothelial deposition of apolipoprotein B (apoB)-containing lipoproteins such as low-density lipoprotein (LDL), which undergo oxidative modification (ox-LDL) to trigger endothelial dysfunction and immune activation.[1,2]
In this framework, disease progression is not merely a matter of plaque volume but of plaque composition and stability. The journey from initial endothelial injury to a life-threatening myocardial infarction involves a complex interplay of lipid deposition, macrophage infiltration, phenotypic switching of vascular smooth muscle cells (VSMCs), and the eventual formation of a necrotic core.[1,3] The final clinical manifestation—whether stable angina or an acute coronary syndrome (ACS)—is dictated by the mechanical integrity of the plaque’s fibrous cap and the biological state of its calcification.[4,9]
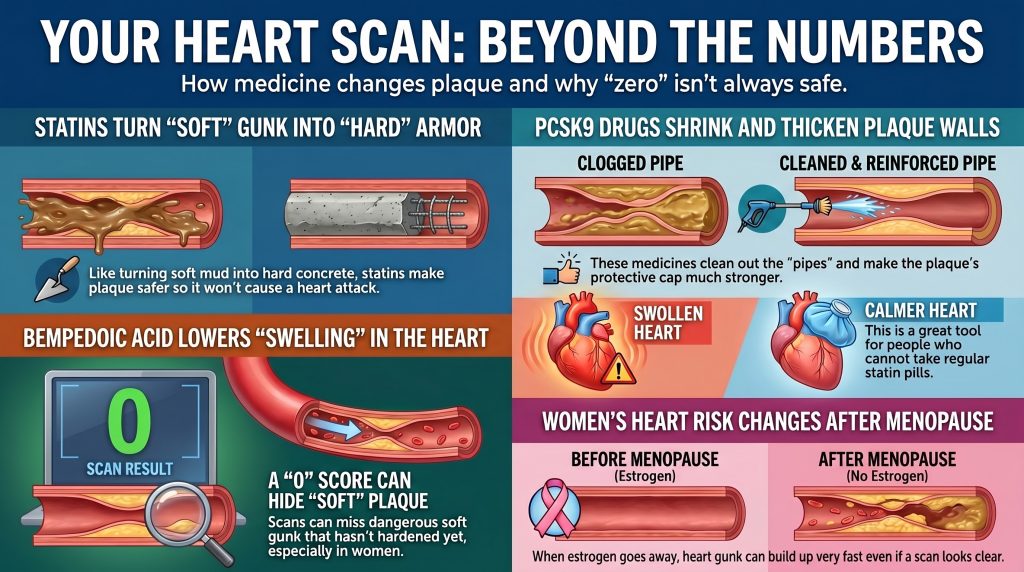
The standard non-invasive tool for assessing atherosclerotic burden has long been the coronary artery calcium (CAC) score. The Agatston score is one of the most powerful predictors of future cardiovascular events, yet its relationship with pharmacological intervention is more complex than it first appears.[5] The emergence of intensive lipid-lowering therapies—including high-intensity statins, PCSK9 inhibitors, and bempedoic acid—has highlighted meaningful differences in how these agents influence plaque biology and radiographic calcification, with important implications for how CAC scores are interpreted in treated patients.[5,9,13]
The Statin–CAC Paradox: Plaque Stabilization and Calcium Densification
Observational cohorts have frequently shown that statin users have higher Agatston scores over time compared with matched untreated individuals, even as their rate of clinical events falls markedly—a pattern sometimes called the “statin–CAC paradox.”[5,7] Randomized controlled trial data on this question are less consistent: heterogeneous CAC quantification methods and limited follow-up durations mean that RCTs have not demonstrated a statistically significant overall effect of statins on CAC progression.[7] The strongest and best-supported imaging signal is not blanket acceleration of calcification but a shift in the nature of the calcium itself: statins are associated with decreases in low-attenuation and fibro-fatty plaque components alongside greater progression of high-density calcium phenotypes.[5,6,45]
Mechanistic Basis of Plaque Stabilization
The “healing hypothesis” proposes that intensive lipid lowering transforms unstable, lipid-rich, inflamed plaques into more fibrotic and mechanically stable lesions.[5,8] As lipid and necrotic components are resolved, the vessel wall undergoes remodeling characterized by deposition of dense calcium that serves as structural reinforcement around the necrotic core, reducing mechanical stress on the fibrous cap and sequestering pro-thrombotic material from the bloodstream.[8]
Serial CCTA data from the PARADIGM study support this model: statin therapy was associated with reductions in low-attenuation and fibro-fatty plaque and with greater progression of high-density calcified plaque, while the increase in overall calcified plaque volume was attenuated in lesions without lipid-rich components.[45] A 2025 serial noncontrast CT study similarly found a statin-associated shift toward denser calcium strata in asymptomatic individuals.[6] These findings provide a more precise characterization than simply stating that statins accelerate CAC progression.
In some patients, therefore, a rising Agatston score under effective lipid-lowering may partly reflect plaque maturation and densification rather than new atherogenesis. However, serial CAC alone cannot reliably distinguish healing from ongoing disease activity on an individual-patient basis, and this interpretation should not be generalized without supporting clinical context.[5]
Proposed Mechanisms: Vitamin K2 and Macrophage Pathways
Several mechanisms have been proposed to explain statin-associated calcium densification. One hypothesis involves inhibition of Vitamin K2 synthesis: Vitamin K2 is a cofactor for Matrix Gla-protein (MGP), a physiological inhibitor of vascular calcification, and reduced Vitamin K2 bioavailability could theoretically remove a brake on the calcification process.[7,8] Macrophage-mediated pathways that normally inhibit calcium deposition may also be suppressed. These remain mechanistic hypotheses drawn largely from preclinical work; they have not been established as dominant pathways in randomized human studies, and the statin-calcification literature explicitly frames this biology as still-disputed.[7,8]
Table 1: Plaque Characteristics: Untreated Progression vs. Statin-Associated Remodeling
| Feature | Untreated Disease Progression | Statin-Associated Remodeling |
| Plaque Composition | High lipid content, large necrotic core | Fibrotic, reduced lipid-rich components |
| Calcium Phenotype | Spotty, low-density microcalcification | Associated shift toward denser, more coalescent calcium strata |
| Fibrous Cap | Thin, inflamed, rupture-prone | Thickened, stable, reduced mechanical stress |
| Agatston Score Trend | Rising due to new lesion formation | May rise due to calcium densification; requires careful clinical interpretation |
| Clinical Risk | Highly elevated | Significantly reduced despite possible calcium score rise |
PCSK9 Inhibitors: Plaque Regression and Fibrous Cap Restoration on Background Statin Therapy
PCSK9 inhibitors such as evolocumab and alirocumab achieve profound reductions in LDL-C by preventing hepatic degradation of low-density lipoprotein receptors (LDLR).[10,11] The principal plaque-imaging trials demonstrated meaningful regression of atheroma volume and improvement in plaque morphology incremental to background statin therapy. Throughout this section it is important to note that GLAGOV, HUYGENS, and PACMAN-AMI all studied PCSK9 inhibition added to statins; their findings cannot be straightforwardly attributed to PCSK9 inhibition as a standalone drug-class effect. Direct evidence on CAC progression with PCSK9 inhibitors is limited and less mature than plaque-regression evidence from IVUS and OCT trials; available data include a small randomized secondary analysis of alirocumab-plus-statin therapy showing attenuated CAC progression versus standard statin-based care, but no large dedicated CAC progression trial comparable to GLAGOV or HUYGENS has been reported.[14,15]
Plaque Regression and Fibrous Cap Thickening
The GLAGOV trial demonstrated that evolocumab added to statin therapy produced significant regression of coronary atheroma volume: a −0.95% nominal change in percent atheroma volume (PAV) versus a +0.05% increase in the placebo group (P<0.001), with 64.3% of evolocumab-treated patients demonstrating PAV regression versus 47.3% in the placebo group (P<0.001). Mean achieved LDL-C was 36.6 mg/dL versus 93.0 mg/dL in the placebo group.[14]
The HUYGENS trial, using serial optical coherence tomography (OCT) following acute myocardial infarction, showed that evolocumab produced significantly greater minimum fibrous cap thickness increase (+42.7 μm vs. +21.5 μm; P=0.015) and maximum lipid arc reduction (−57.5° vs. −31.4°; P=0.04) compared with placebo.[15]
The PACMAN-AMI trial demonstrated that alirocumab initiated within 24 hours of acute myocardial infarction produced significantly greater regression in percent atheroma volume (PAV −2.13% vs. −0.92%, P<0.001) and greater reduction in normalized total atheroma volume (−26.12 mm³ vs. −14.97 mm³), as well as greater fibrous cap thickening, compared with statin monotherapy.[11]
Vascular Biology: Established and Emerging Signals
Preclinical studies have identified potential LDLR-independent effects of PCSK9 on atherogenesis, including inhibition of monocyte adhesion to stimulated human coronary artery endothelial cells (HCAECs) via the VCAM-1 and ICAM-1 pathways.[12,16] In large cardiovascular outcome trials, however, PCSK9 inhibition has generally shown minimal change in hsCRP, indicating that the clinically established primary mechanism of benefit is profound apoB/LDL lowering rather than a systemic anti-inflammatory effect. Pleiotropic or local vascular effects suggested by preclinical work may contribute, but this should not be overstated given the largely neutral hsCRP signal in human trial data.[17]
Lipoprotein(a): An Important Independent Risk Factor
A meaningful advantage of PCSK9 inhibitors is their ability to lower Lipoprotein(a) [Lp(a)], an independent and genetically determined cardiovascular risk factor largely unaffected by statin therapy.[10] PCSK9 inhibitors reduce Lp(a) by approximately 25–27% (median 26.9% in the FOURIER evolocumab cohort). The relationship between Lp(a) levels and CAC burden is inconsistent across studies; Lp(a) and CAC appear to be independently informative risk markers rather than tightly correlated measures. Clinicians should therefore distinguish the robust evidence that elevated Lp(a) is an independent ASCVD risk factor from the weaker and less consistent evidence that Lp(a) directly predicts CAC burden.[10,17]
Bempedoic Acid: Proven Outcomes, Limited Plaque Imaging Evidence
Bempedoic acid is an oral ATP citrate lyase (ACL) inhibitor that acts upstream of HMG-CoA reductase in the cholesterol biosynthesis pathway.[19,23] As a prodrug, it is converted into its active metabolite, bempedoyl-CoA, by the enzyme very long-chain acyl-CoA synthetase 1 (ACSVL1). This activating enzyme is expressed in the liver but is absent in skeletal muscle, providing the mechanistic basis for the drug’s low incidence of muscle-related adverse effects and making it a valuable option for statin-intolerant patients.[20,23]
Cardiovascular Outcomes: The CLEAR Trials
The CLEAR Outcomes trial established clinical efficacy in high-risk, statin-intolerant patients.[21] Over a median follow-up of 3.4 years, bempedoic acid achieved a 21.1% placebo-corrected reduction in LDL-C and a statistically significant 13% relative risk reduction in the primary MACE-4 endpoint [HR 0.87, 95% CI 0.79–0.96; P=0.004].[21] A prespecified total-event analysis demonstrated a 20% reduction in total MACE-4 events [HR 0.80, 95% CI 0.72–0.89; P<0.001].[22]
Table 2: CLEAR Outcomes Trial: Cardiovascular Endpoint Summary (Nissen et al., NEJM 2023; JAMA 2024)
| Outcome (CLEAR Outcomes) | HR | 95% CI |
| MACE-4 — Primary Endpoint | 0.87 | 0.79–0.96 (P = 0.004) |
| MACE-3 (CV Death / MI / Stroke) | 0.85 | 0.76–0.96 |
| Fatal or Nonfatal MI | 0.77 | 0.66–0.91 |
| Coronary Revascularization | 0.81 | 0.72–0.92 |
| Total MACE-4 Events (prespecified) | 0.80 | 0.72–0.89 (P < 0.001) |
Inflammation, Mechanism, and Imaging Evidence
A distinguishing feature of bempedoic acid is its consistent reduction of hsCRP: a 22% placebo-corrected median reduction was observed at 6 months in CLEAR Outcomes.[19,21] Activation of the AMP-activated protein kinase (AMPK) pathway has been proposed as a contributing cellular mechanism, based primarily on preclinical and mechanistic review data.[20,23] More recent work suggests the drug’s biology may be more complex than initially characterized, with preliminary evidence of potential direct PPARα activation; these mechanistic questions remain under active investigation and should not be presented as settled biology.
Bempedoic acid has proven outcomes benefit and hsCRP lowering in statin-intolerant patients, but its direct effects on coronary plaque morphology remain largely unproven pending dedicated imaging trials.[24] The only published plaque-imaging evidence is a single case report describing shrinkage of low-attenuation plaque with long-term bempedoic acid monotherapy—a hypothesis-generating observation, not proof of a plaque-regression class effect. A phase 3 imaging trial registered in 2026 evaluates the combination of bempedoic acid, ezetimibe, and high-intensity statin therapy on coronary plaque; its design tests triple combination therapy rather than bempedoic acid in isolation, and results have not yet been reported.
Limitations and Blind Spots of CAC = 0: Moving Beyond a Binary Interpretation
The observation that a CAC score of 0 predicts very low 10-year cardiovascular risk—the “power of zero”—has established CAC as a useful gatekeeper in risk stratification.[25,27,29] CAC = 0 is a genuinely powerful short-term negative risk marker in most clinical settings. However, it has important and well-characterized blind spots that require careful interpretation, particularly in symptomatic patients, younger individuals, and women in mid-life.[30]
Non-Calcified Plaque: The Underdetected Component
A CAC score of 0 does not exclude coronary artery disease. Non-calcified plaque (NCP), which is lipid-rich and highly vulnerable to rupture, is entirely invisible on non-contrast CT.[25,28] Studies using CCTA have shown that approximately 15–17% of low-to-intermediate-risk individuals with CAC = 0 have detectable atherosclerosis, with roughly 3.5% harboring obstructive disease (>50% stenosis) despite a zero calcium score.[28,30]
In a diverse symptomatic Montefiore cohort analyzed by Rozanski et al., a zero CAC score retained very high negative predictive value for obstructive coronary disease and near-term adverse events, and only a small percentage of CAC = 0 patients—approximately 7%—had detectable noncalcified plaque on CCTA.[30] Separate prospective data from stable-chest-pain populations likewise show very low MACE event rates when CAC = 0, though obstructive CAD is not completely excluded. These data together confirm that CAC = 0 is a genuinely powerful short-term reassurance signal, while still leaving a clinically meaningful minority with non-calcified plaque that calcium scoring cannot detect.
The mechanisms of clinical events in individuals with CAC = 0 typically involve rupture or erosion of non-calcified lesions, systemic inflammation, or coronary microvascular dysfunction.[27,31] Plaque erosion—disruption of the endothelial surface over a lipid-rich non-calcified plaque—is particularly common in women and younger individuals.[27]
Prevalence Estimates Depend Critically on Cohort and Method
In the SCAPIS (Swedish Cardiopulmonary Bioimage Study), among middle-aged adults from the general population with CAC = 0, approximately 5.5% had any coronary plaque detectable on CCTA and 0.4% had significant stenosis.[29] The frequently cited 9.2% plaque-prevalence figure from SCAPIS applies specifically to the subgroup with CAC = 0 and intermediate 10-year ASCVD risk—an important distinction, because applying this conditional estimate to the entire CAC-zero general population would materially overstate disease prevalence.[29]
AI-QCT registries of symptomatic patients referred for clinical CCTA report substantially higher plaque detection rates in CAC = 0 individuals—ranging from approximately 54% to 95% in some selected cohorts.[26] These figures are not generalizable to screening populations; they reflect the higher pre-test probability of clinical referral cohorts as well as variation in AI-QCT protocols, thresholds, and definitions across studies. Prevalence estimates from symptomatic, asymptomatic, general-population, and AI-QCT registry studies should not be combined without careful population-specific stratification.[30]
Table 3: Prevalence of Non-Calcified Plaque in CAC = 0 Patients: Context-Dependent Estimates Across Study Populations
| Study / Cohort | Population | Key Finding for CAC = 0 Patients |
| Cheng et al. | Low to intermediate risk, asymptomatic | ~16% with detectable non-calcified plaque on CCTA |
| Rubinshtein et al. | Symptomatic | ~14.5% with plaque; ~3% with obstructive disease |
| Rozanski et al. (JCCT 2023) | Symptomatic stable chest pain, diverse population | ~7% NCP on CCTA; very high NPV for obstructive disease and near-term events; diverse symptomatic Montefiore cohort |
| SCAPIS (Bergström et al.) | Middle-aged general population | ~5.5% any plaque overall; ~9.2% specifically in the CAC=0 + intermediate-risk subgroup [29] |
| AI-QCT registries (selected clinical CCTA cohorts) | Symptomatic / high pre-test probability | 54–95%: highly dependent on cohort selection, referral indication, and AI-QCT protocol. Not generalizable to screening populations [26] |
Sex-Specific Cardiovascular Risk and Plaque Biology
Women exhibit important differences in the presentation and pathophysiology of atherosclerotic cardiovascular disease (ASCVD) compared with men. Women generally develop clinically manifest coronary artery disease approximately a decade later than men, a phenomenon largely attributed to the vasoprotective effects of endogenous estrogen.[37,39]
The Protective Role of Estrogen and the Menopause Transition
Estrogen maintains vascular health through multiple pathways: it promotes endothelium-dependent vasodilation by stimulating nitric oxide (NO) and prostacyclin synthesis, inhibits vascular smooth muscle cell contraction and proliferation, and exerts anti-inflammatory effects on the vessel wall.[35,38] Estrogen also maintains more favorable lipid profiles throughout the premenopausal years.[35]
The menopause transition is associated with a precipitous decline in circulating estradiol, triggering accelerated vascular aging. This shift produces measurable reductions in endothelial function as assessed by flow-mediated dilation (FMD), increased systemic oxidative stress, and progressive worsening of arterial stiffness, blood pressure, and atherogenic dyslipidemia.[34,36] FMD in late postmenopausal women is approximately 50% lower than in premenopausal women of similar age, with the most significant decline occurring during the perimenopausal transition.[34]
Risk Acceleration After Menopause
As women enter their 60s, cardiovascular risk accelerates markedly, with CVD incidence approaching that of age-matched men.[37,38] Longitudinal data from the Healthy Women Study show that among postmenopausal women with a baseline CAC of 0, approximately 33% develop new, incident calcification over a subsequent 6-year period, with premenopausal LDL-C and HDL-C as strong predictors.[39]
Early Menopause and Long-Term Risk
Women who experience early menopause (EM, defined as menopause before age 45) face substantially greater long-term cardiovascular risks. In the Multi-Ethnic Study of Atherosclerosis (MESA), more than half of postmenopausal women with early menopause had a baseline CAC score of 0.[40] Among those women with CAC = 0, 10-year ASCVD incidence was low-to-borderline, yet those with early menopause had significantly higher 15-year risk than those without (adjusted HR 1.96, 95% CI 1.26–3.04).[40] This illustrates the core diagnostic problem: the same CAC = 0 result that appears reassuring at 10 years can mask a substantially elevated long-term risk trajectory in women with early menopause, driven by prolonged estrogen deficiency that accelerates subclinical plaque accumulation after the initial scan.
Systematic meta-analyses confirm that early menopause is independently associated with a 50–60% increase in lifetime coronary heart disease risk, an effect only partially attenuated by traditional risk factor adjustment.[41]
Sex Differences in Plaque Morphology and Detection
Women generally have lower absolute CAC burden than men and, in several cohorts, lower absolute plaque burden as well; however, recent AI-QCT data suggest that the relative cardiovascular risk associated with a given increase in plaque burden is greater in women than in men.[27,33] Women are significantly more likely to develop non-calcified, lipid-rich plaques prone to endothelial erosion rather than fibrous cap rupture—yet both mechanisms trigger MACE. Because CAC scoring captures only the calcified component of disease, it may be a less complete risk indicator in women, who often carry a greater proportion of their total plaque burden in non-calcified form. Women with any detectable CAC face disproportionately higher relative cardiovascular risk: CAC Consortium data show approximately 1.3-fold higher hazard of cardiovascular death in women than in men at equivalent CAC burden.[27]
In their 40s and 50s, women may accumulate a substantial burden of lipid-rich NCP that will only become radiographically detectable as calcium after the post-menopausal acceleration of vascular calcification.[27,33] This creates a diagnostic window during midlife when traditional risk factors are rising and inflammatory plaque is accumulating, yet CAC remains at zero. The absence of calcium may be an especially incomplete risk signal precisely in this population—younger, perimenopausal women—where near-term absolute risk appears lowest but medium-term risk may be substantially higher than the calcium score suggests.
Table 4: Sex-Specific Cardiovascular Risk per Unit Plaque Volume: Selected Findings from the CONFIRM2 Registry (Choi et al., Circ Cardiovasc Imaging 2025 [33])
| Finding — CONFIRM2 Registry (Choi et al. 2025 [33]) | Women | Men |
| Relative risk increase per 50 mm³ total plaque volume | +17.7% (RR ≈1.18) | +5.3% (RR ≈1.05) |
| Pattern across NCP, calcified plaque, and high-risk plaque subtypes | Larger relative risk increments across all subtypes | Smaller relative risk increments across all subtypes |
| Principal published conclusion | Substantially higher relative cardiovascular risk per unit plaque | Lower relative risk per equivalent plaque unit |
The total plaque volume risk increment (+17.7% per 50 mm³ in women vs +5.3% in men) is reported from the published CONFIRM2 results. The directional pattern across non-calcified, calcified, and high-risk plaque subtypes (larger increments in women) is the principal published conclusion of that paper; exact subtype hazard ratios should be transcribed from the published results table before any submission requiring those specific values.
Advanced Imaging: The Role of AI-Enhanced CCTA
Artificial intelligence–based quantitative CT (AI-QCT) represents a meaningful advance in non-invasive cardiovascular risk assessment. By automating measurement of total plaque burden—including calcified, non-calcified, and low-attenuation components—AI-QCT provides a more comprehensive characterization of disease than traditional CAC scoring alone.[26,33] These tools are rapidly evolving, but broad standardization across institutions and vendors, external validation in diverse populations, sex-specific risk thresholds, and integration into clinical guidelines are still ongoing processes. AI-QCT is best understood as a promising emerging modality rather than a fully standardized replacement for current CAC and CCTA frameworks.
AI-QCT, CAC, and Reclassification in Symptomatic Cohorts
In symptomatic patients referred for clinical CCTA, research demonstrates only moderate categorical agreement between traditional CAC scores and AI-derived total plaque burden, with discordance particularly pronounced in women.[42] AI-QCT frequently identifies significant NCP in patients with zero Agatston scores in these referral populations. Prevalence varies widely by cohort, scanner protocol, and AI-QCT algorithm, and figures from clinical CCTA registries should not be extrapolated to general screening populations. Nonetheless, the reclassification is clinically meaningful for symptomatic patients in whom a zero calcium score might otherwise prematurely terminate the diagnostic workup, especially women with additional risk features.[26,30,42]
AI-QCT further enables quantification of high-risk plaque (HRP) features including low-attenuation plaque (LAP) and positive coronary remodeling. Data from the CONFIRM2 registry demonstrate that these features carry significantly higher relative risk in women than in men per unit of plaque volume, consistent with the clinical observation that women’s smaller-caliber coronary vessels may render each unit of high-risk plaque more hemodynamically consequential.[33]
Integrating Epicardial Adipose Tissue Analysis
Automated analysis of epicardial adipose tissue (EAT) volume and attenuation from routine calcium-scoring CT scans has been shown to significantly enhance MACE prediction, with particularly strong incremental value in women.[43] EAT is metabolically active and exerts paracrine pro-inflammatory and pro-atherogenic effects on adjacent coronary arteries through the secretion of cytokines and adipokines,[32] and integrating quantitative EAT features with CAC scoring and conventional risk factors provides a more biologically comprehensive sex-specific risk assessment than calcified plaque metrics alone, though this approach requires further standardization and prospective validation before routine clinical implementation.[43]
An Integrated Framework for Therapeutic Mapping
A useful framework maps atherosclerosis progression through specific biological stages and identifies where different therapeutic classes exert their primary mechanistic influence. This mapping should be understood as directionally plausible based on available mechanistic and imaging data, not as a clinically proven stage-by-stage model validated by head-to-head drug-class trials.[1,4,9]
Stages of Plaque Development and Therapeutic Positioning
Stage 1 — Endothelial Activation: Endothelial dysfunction, reduced NO bioavailability, and subendothelial retention of apoB-containing lipoproteins. This stage is substantially modulated by endogenous estrogen and is the proposed target of early PCSK9 inhibition based on preclinical evidence.[2,12]
Stage 2 — Early Non-Calcified Plaque: Macrophage foam cell formation establishes the earliest atheromatous lesion. Bempedoic acid and statins reduce the circulating lipid pool available for subendothelial accumulation and dampen systemic inflammation.[1,20]
Stage 3 — Intermediate / Mixed Plaque: Extracellular lipid accumulation and microcalcifications emerge within necrotic core zones. High-intensity statins and PCSK9i (when added to statins) drive plaque regression and fibrous cap maturation, with the strongest direct human trial evidence at this stage.[1,9,14]
Stage 4 — Fibrocalcific / Stable Plaque: Dense, coalescent calcification with a thick collagen-rich fibrous cap, representing the histopathological end-state of healed plaque, associated primarily with intensive statin therapy and the calcium densification process.[5,8,9]
Comparative Therapeutic Profile
Statins are best characterized as plaque stabilizers associated with calcium densification in observational and serial CCTA data. They have the strongest and broadest evidence base for reducing lipid-rich plaque and promoting transformation of vulnerable lesions toward denser, more stable phenotypes. A rising Agatston score under statin therapy should be interpreted in the context of documented densification and clinical improvement rather than as evidence of therapeutic failure.[5,7,8,45]
PCSK9 Inhibitors are best characterized as plaque regressionors and fibrous cap restorers when added to statin therapy in high-risk imaging cohorts. Their principal established mechanism of clinical benefit is profound apoB/LDL lowering, with meaningful incremental plaque regression and cap thickening beyond statins alone. Pleiotropic effects suggested by preclinical data have not translated into a clear systemic anti-inflammatory signal in human outcome trials.[11,12,14,15]
Bempedoic Acid functions as an upstream lipid-lowering and hsCRP-lowering agent with proven outcomes benefit in statin-intolerant patients. Its plaque-modification effects are mechanistically plausible but supported by very limited imaging data; robust serial plaque imaging evidence has not yet been reported.[19,20,21]
Clinical Implications and Guideline Directions
The integration of pharmacologic mechanisms, advanced imaging, and sex-specific plaque biology highlights important gaps in current clinical practice and supports a shift toward biologically informed, individualized risk assessment.
Interpreting CAC = 0 in Symptomatic and Mid-Life Women
The cumulative evidence supports a calibrated interpretation of CAC = 0: it is a powerful short-term negative risk marker that should not be dismissed, yet it should not automatically end risk assessment in younger or symptomatic women—especially in the presence of early menopause, strong family history, diabetes, elevated Lp(a), or persistent inflammatory risk.[27,40,41] A CAC of 0 in a woman with these characteristics may correctly predict low 10-year event risk while substantially underestimating 15-year risk driven by ongoing non-calcified plaque accumulation and the biological effects of prolonged estrogen deficiency. In these settings, a CAC of 0 should invite further characterization of risk rather than conclude risk stratification.
A More Comprehensive Approach to Sex-Conscious Risk Assessment
A multi-modal, sex-conscious risk assessment framework incorporates several complementary elements.
Sex-specific risk factors—including age at menopause, duration of estrogen deficiency, and reproductive history—should be formally integrated into clinical risk discussions, as they carry independent prognostic weight beyond traditional Framingham-based variables.[37,41]
Advanced imaging via CCTA should be considered when CAC scoring may underestimate true atherosclerotic burden, particularly in symptomatic women with CAC = 0 and additional risk features. The 2024 ESC Guidelines for the Management of Chronic Coronary Syndromes recommend first-line noninvasive anatomic or functional imaging for suspected CCS, selected according to pretest likelihood, patient characteristics, and local expertise. Within that framework, CCTA is specifically preferred for ruling out obstructive CAD and identifying nonobstructive disease, and the guidelines explicitly incorporate age, sex, and symptom character into pretest likelihood models.[26,31,33]
Integrated biomarker-imaging models combining Lp(a), hsCRP, and emerging EAT fat-omics analysis represent a biologically more complete framework for sex-specific risk stratification. These approaches require further standardization and prospective validation before routine implementation.[43]
Limitations and Open Questions
The evidence reviewed here has several important limitations that must be recognized when applying these concepts clinically or in academic scholarship.
Most plaque-imaging evidence for PCSK9 inhibitors derives from studies of add-on therapy to background statins, not from clean mechanistic comparisons between drug classes. GLAGOV, HUYGENS, and PACMAN-AMI do not establish what PCSK9 inhibitors achieve independently of statins, and their imaging endpoints cannot be directly compared with statin-only or bempedoic acid-only imaging data.[11,14,15]
The bempedoic acid plaque-imaging literature currently consists of a single case report and one recently registered phase 3 imaging trial that has not yet reported results. Outcomes-based conclusions about bempedoic acid are well supported; morphologic plaque-imaging conclusions are not.[24]
AI-QCT plaque detection rates vary substantially by cohort selection, referral indication, and AI-QCT protocol. Plaque prevalence figures from symptomatic CCTA referral populations should not be extrapolated to general screening populations. Standardized thresholds, validated sex-specific risk cutoffs, and prospective outcome data for AI-QCT remain incomplete.[26,33,42]
The field still lacks clean head-to-head randomized trials designed to compare drug class effects on CAC progression or plaque morphology using sex-stratified pre-specified endpoints and the same imaging platform. The mechanistic pathways proposed for statin-associated calcium densification (Vitamin K2 inhibition, macrophage suppression) and for bempedoic acid’s anti-inflammatory effects (AMPK, possible PPARα activation) are supported primarily by preclinical evidence and remain incompletely validated in human studies.[7,8,20,23]
Conclusion
Intensive apoB-lowering therapy reduces cardiovascular risk across drug classes, but its imaging footprints differ in important ways. Statins have the strongest and longest-established evidence for reducing lipid-rich plaque and promoting a denser, more stable calcium phenotype; their association with rising Agatston scores is best understood as calcium densification rather than uncomplicated disease progression. PCSK9 inhibitors provide additional plaque regression, lipid arc reduction, and fibrous cap thickening when added to statins, with a primarily LDL-lowering mechanism driving outcome benefit rather than a systemic anti-inflammatory one. Bempedoic acid offers proven event reduction and meaningful hsCRP lowering in statin-intolerant patients, but its effects on coronary plaque morphology remain largely unproven pending dedicated imaging trials.
CAC remains a powerful marker of calcified plaque burden and near-term prognosis, but it incompletely captures non-calcified atherosclerosis—particularly in symptomatic patients and women in mid-life. CAC = 0 should be interpreted as one component of a biologically informed risk assessment rather than as a stand-alone arbiter of disease absence. In most clinical contexts it provides powerful short-term reassurance; in specific contexts—early menopause, symptomatic presentations, elevated Lp(a), family history, or persistent inflammatory risk—it should prompt further rather than conclude risk stratification.
For women specifically, the distinct biology of plaque formation—characterized by early lipid-rich non-calcified lesions, a sensitive period of post-menopausal risk acceleration, and significantly higher relative cardiovascular risk per unit of plaque volume—demands an individualized approach to imaging and lipid-lowering therapy. A sex-conscious, multi-modal risk assessment framework incorporating CCTA, sex-specific biomarkers, and comprehensive clinical context offers the most evidence-consistent path toward equitable and effective cardiovascular disease prevention.
References
- Libby P. Inflammation in atherosclerosis. Nature. 2002;420(6917):868-874. doi:10.1038/nature01323
- Han Y, Xu H, Yao X, et al. Molecular mechanisms and therapeutic progress in atherosclerosis: bridging immune inflammation and precision medicine. Front Immunol. 2026;16:1737662. Published 2026 Jan 5. doi:10.3389/fimmu.2025.1737662
- Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145(3):341-355. doi:10.1016/j.cell.2011.04.005
- Papafaklis MI, Koros R, Tsigkas G, Karanasos A, Moulias A, Davlouros P. Reversal of Atherosclerotic Plaque Growth and Vulnerability: Effects of Lipid-Modifying and Anti-Inflammatory Therapeutic Agents. Biomedicines. 2024;12(11):2435. Published 2024 Oct 23. doi:10.3390/biomedicines12112435
- Khanna S, Nerlekar N, Bhat A. Reconciling Coronary Artery Calcification in the Lipid-Lowering Era. JACC Adv. 2026;5(1):102506. doi:10.1016/j.jacadv.2025.102506
- Giovannucci J, Shanbhag A, Hong W, et al. Impact of statins on progression of coronary artery calcium composition and density as assessed by noncontrast CT. Int J Cardiovasc Imaging. 2025;41(12):2481-2492. doi:10.1007/s10554-025-03561-0
- Shahraki MN, Jouabadi SM, Bos D, Stricker BH, Ahmadizar F. Statin Use and Coronary Artery Calcification: a Systematic Review and Meta-analysis of Observational Studies and Randomized Controlled Trials. Curr Atheroscler Rep. 2023;25(11):769-784. doi:10.1007/s11883-023-01151-w
- Kadoglou NP, Stasinopoulou M, Velidakis N, et al. The Complex Mechanisms and the Potential Effects of Statins on Vascular Calcification: A Narrative Review. Rev Cardiovasc Med. 2024;25(2):51. Published 2024 Jan 30. doi:10.31083/j.rcm2502051
- Papafaklis MI, Koros R, Tsigkas G, Karanasos A, Moulias A, Davlouros P. Reversal of Atherosclerotic Plaque Growth and Vulnerability: Effects of Lipid-Modifying and Anti-Inflammatory Therapeutic Agents. Biomedicines. 2024;12(11):2435. Published 2024 Oct 23. doi:10.3390/biomedicines12112435
- Wu Z, Gao L, Lin Z. Can proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors regress coronary atherosclerotic plaque? A systematic review and meta-analysis. Am J Transl Res. 2023;15(1):452-465. Published 2023 Jan 15.
- Räber L, Ueki Y, Otsuka T, et al. Effect of Alirocumab Added to High-Intensity Statin Therapy on Coronary Atherosclerosis in Patients With Acute Myocardial Infarction: The PACMAN-AMI Randomized Clinical Trial. JAMA. 2022;327(18):1771-1781. doi:10.1001/jama.2022.5218
- Zulkapli R, Muid SA, Wang SM, Nawawi H. PCSK9 Inhibitors Reduce PCSK9 and Early Atherogenic Biomarkers in Stimulated Human Coronary Artery Endothelial Cells. Int J Mol Sci. 2023;24(6):5098. Published 2023 Mar 7. doi:10.3390/ijms24065098
- Ahmadi A, Argulian E, Leipsic J, Newby DE, Narula J. From Subclinical Atherosclerosis to Plaque Progression and Acute Coronary Events: JACC State-of-the-Art Review. J Am Coll Cardiol. 2019;74(12):1608-1617. doi:10.1016/j.jacc.2019.08.012
- Nicholls SJ, Puri R, Anderson T, et al. Effect of Evolocumab on Progression of Coronary Disease in Statin-Treated Patients: The GLAGOV Randomized Clinical Trial. JAMA. 2016;316(22):2373-2384. doi:10.1001/jama.2016.16951
- Nicholls SJ, Kataoka Y, Nissen SE, et al. Effect of Evolocumab on Coronary Plaque Phenotype and Burden in Statin-Treated Patients Following Myocardial Infarction. JACC Cardiovasc Imaging. 2022;15(7):1308-1321. doi:10.1016/j.jcmg.2022.03.002
- Barbieri L, Tumminello G, Fichtner I, et al. PCSK9 and Coronary Artery Plaque-New Opportunity or Red Herring?. Curr Atheroscler Rep. 2024;26(10):589-602. doi:10.1007/s11883-024-01230-6
- Pradhan AD, Aday AW, Rose LM, Ridker PM. Residual Inflammatory Risk on Treatment With PCSK9 Inhibition and Statin Therapy. Circulation. 2018;138(2):141-149. doi:10.1161/CIRCULATIONAHA.118.034645
- Chen H, Chen X. PCSK9 inhibitors for acute coronary syndrome: the era of early implementation. Front Cardiovasc Med. 2023;10:1138787. Published 2023 May 2. doi:10.3389/fcvm.2023.1138787
- Jia X, Virani SS. CLEAR Serenity Trial: More Clarity for the Future of Bempedoic Acid in Patients Unable to Take Statins?. J Am Heart Assoc. 2019;8(7):e012352. doi:10.1161/JAHA.119.012352
- Pinkosky SL, Newton RS, Day EA, et al. Liver-specific ATP-citrate lyase inhibition by bempedoic acid decreases LDL-C and attenuates atherosclerosis. Nat Commun. 2016;7:13457. Published 2016 Nov 28. doi:10.1038/ncomms13457
- Nissen SE, Lincoff AM, Brennan D, et al. Bempedoic Acid and Cardiovascular Outcomes in Statin-Intolerant Patients. N Engl J Med. 2023;388(15):1353-1364. doi:10.1056/NEJMoa2215024
- Nicholls SJ, Nelson AJ, Lincoff AM, et al. Impact of Bempedoic Acid on Total Cardiovascular Events: A Prespecified Analysis of the CLEAR Outcomes Randomized Clinical Trial. JAMA Cardiol. 2024;9(3):245-253. doi:10.1001/jamacardio.2023.5155
- Biolo G, Vinci P, Mangogna A, et al. Mechanism of action and therapeutic use of bempedoic acid in atherosclerosis and metabolic syndrome. Front Cardiovasc Med. 2022;9:1028355. Published 2022 Oct 28. doi:10.3389/fcvm.2022.1028355
- Korosoglou G, Giesen A, Geiss E, Stach K. Case report: Strong low-density-cholesterol reduction accompanied by shrinkage of low-attenuation coronary plaque during lipid-lowering treatment with bempedoic acid-serial evaluation by coronary computed tomography angiography. Front Cardiovasc Med. 2023;10:1203832. Published 2023 Aug 4. doi:10.3389/fcvm.2023.1203832
- Koulaouzidis G, Charisopoulou D, Jenkins PJ, Koulaouzidis A, McArthur T. Prevalence of noncalcified coronary plaque in patients with calcium score of 0: the silent enemy. Angiology. 2013;64(3):205-210. doi:10.1177/0003319712440618
- Khan NA, Wesbey III G, Cobb G, et al. Using AI-Quantitative CT to evaluate the relationship between coronary artery calcium and segment involvement scores in quantifying coronary plaque burden. Int J Cardiovasc Imaging. 2026;42(1):49-59. doi:10.1007/s10554-025-03569-6
- Shaw LJ, Min JK, Nasir K, et al. Sex differences in calcified plaque and long-term cardiovascular mortality: observations from the CAC Consortium. Eur Heart J. 2018;39(41):3727-3735. doi:10.1093/eurheartj/ehy534
- Cho I, Suh JW, Chang HJ, et al. Prevalence and prognostic implication of non-calcified plaque in asymptomatic population with coronary artery calcium score of zero. Korean Circ J. 2013;43(3):154-160. doi:10.4070/kcj.2013.43.3.154
- Bergström G, Persson M, Adiels M, et al. Prevalence of Subclinical Coronary Artery Atherosclerosis in the General Population. Circulation. 2021;144(12):916-929. doi:10.1161/CIRCULATIONAHA.121.055340
- Fattouh M, Kuno T, Pina P, et al. Interplay Between Zero CAC, Quantitative Plaque Analysis, and Adverse Events in a Diverse Patient Cohort. Circ Cardiovasc Imaging. 2023;16(8):e015236. doi:10.1161/CIRCIMAGING.123.015236
- Vrints C, Andreotti F, Koskinas KC, et al. 2024 ESC Guidelines for the management of chronic coronary syndromes. Eur Heart J. 2024;45(36):3415-3537. doi:10.1093/eurheartj/ehae177
- Antonopoulos AS, Sanna F, Sabharwal N, et al. Detecting human coronary inflammation by imaging perivascular fat. Sci Transl Med. 2017;9(398):eaal2658. doi:10.1126/scitranslmed.aal2658
- Feuchtner GM, Lacaita PG, Bax JJ, et al. AI-Quantitative CT Coronary Plaque Features Associate With a Higher Relative Risk in Women: CONFIRM2 Registry. Circ Cardiovasc Imaging. 2025;18(6):e018235. doi:10.1161/CIRCIMAGING.125.018235
- Moreau KL, Hildreth KL, Meditz AL, Deane KD, Kohrt WM. Endothelial function is impaired across the stages of the menopause transition in healthy women. J Clin Endocrinol Metab. 2012;97(12):4692-4700. doi:10.1210/jc.2012-2244
- Reslan OM, Khalil RA. Vascular effects of estrogenic menopausal hormone therapy. Rev Recent Clin Trials. 2012;7(1):47-70. doi:10.2174/157488712799363253
- Moreau KL, Hildreth KL, Klawitter J, Blatchford P, Kohrt WM. Decline in endothelial function across the menopause transition in healthy women is related to decreased estradiol and increased oxidative stress. Geroscience. 2020;42(6):1699-1714. doi:10.1007/s11357-020-00236-7
- Mishra T, Pasnoor DS, Gandhi M, et al. Interplay of menopause, coronary artery calcium score and cardiovascular disease risk. World J Cardiol. 2025;17(11):109627. doi:10.4330/wjc.v17.i11.109627
- SenthilKumar G, Katunaric B, Bordas-Murphy H, Sarvaideo J, Freed JK. Estrogen and the Vascular Endothelium: The Unanswered Questions. Endocrinology. 2023;164(6):bqad079. doi:10.1210/endocr/bqad079
- Kuller LH, Matthews KA, Edmundowicz D, Chang Y. Incident coronary artery calcium among postmenopausal women. Atherosclerosis. 2008;200(2):278-285. doi:10.1016/j.atherosclerosis.2007.12.057
- Chu JH, Michos ED, Ouyang P, et al. Coronary artery calcium and atherosclerotic cardiovascular disease risk in women with early menopause: The Multi-Ethnic Study of Atherosclerosis (MESA). Am J Prev Cardiol. 2022;11:100362. Published 2022 Jun 13. doi:10.1016/j.ajpc.2022.100362
- Muka T, Oliver-Williams C, Kunutsor S, et al. Association of Age at Onset of Menopause and Time Since Onset of Menopause With Cardiovascular Outcomes, Intermediate Vascular Traits, and All-Cause Mortality: A Systematic Review and Meta-analysis. JAMA Cardiol. 2016;1(7):767-776. doi:10.1001/jamacardio.2016.2415
- Khan NA, Wesbey III G, Cobb G, et al. Using AI-Quantitative CT to evaluate the relationship between coronary artery calcium and segment involvement scores in quantifying coronary plaque burden. Int J Cardiovasc Imaging. 2026;42(1):49-59. doi:10.1007/s10554-025-03569-6
- Singh P, Hu T, Hoori A, Al-Kindi S, Wilson DL, Rajagopalan S. Sex-specific cardiovascular risk prediction using AI-derived epicardial adipose tissue measurements on CT calcium scoring exams. Am J Prev Cardiol. 2025;25:101367. Published 2025 Dec 8. doi:10.1016/j.ajpc.2025.101367
- Narula J, Chandrashekhar Y, Ahmadi A, et al. SCCT 2021 Expert Consensus Document on Coronary Computed Tomographic Angiography: A Report of the Society of Cardiovascular Computed Tomography. J Cardiovasc Comput Tomogr. 2021;15(3):192-217. doi:10.1016/j.jcct.2020.11.001
- Lee SE, Chang HJ, Sung JM, et al. Effects of Statins on Coronary Atherosclerotic Plaques: The PARADIGM Study. JACC Cardiovasc Imaging. 2018;11(10):1475-1484. doi:10.1016/j.jcmg.2018.04.015