1. Introduction: The Cholesterol Mystery
Imagine a man named John. John is 55 years old, active, and diligent about his health. He eats a Mediterranean diet and walks three miles every morning. At his last annual checkup, his doctor delivered what seemed like excellent news: his LDL cholesterol—the “bad” kind—was 80 mg/dL. In the world of standard medicine, that is a gold-star score. John felt invincible.
Two months later, while working in his garden, John suffered a massive heart attack.
How could this happen? If his “bad cholesterol” was low, why did his arteries clog? This is the great mystery that has haunted cardiology for decades. The answer lies in a single lab test that most doctors are not yet ordering: Apolipoprotein B, or ApoB.
To understand why this happens, we have to look at the Evidence Ladder— a tool science journalists use to separate “guesses” from “facts.”
- Tier A (The Gold Standard): Proven by major trials and genetic studies. We are certain about these.
- Tier B: Likely true, based on strong genetic evidence, but still waiting for the final trial results.
- Tier C: Associated with disease, meaning it is a great “warning sign,” but the exact “cause and effect” is still being mapped out.
- Tier D: A fascinating “hunch” that scientists are currently investigating in the lab.
For years, we have been weighing “cholesterol” when we should have been counting the dangerous particles. This blog post is a guide to the 21st-century science of heart health. We are looking beyond the heart to see how one tiny particle affects your brain, your eyes, and your longevity.
2. Takeaway 1: It’s the Number of Trucks, Not the Weight of the Cargo
The biggest mistake in modern medicine is confusing the “cargo” for the “truck.” To understand your risk, you must understand that cholesterol does not float freely in your blood; it is carried inside little “trucks” called lipoproteins.
The standard LDL-C test measures the total weight of the cholesterol (the cargo). ApoB, however, measures the number of trucks. This is critical because every single dangerous particle—whether it is an LDL, a VLDL, or a remnant particle—has exactly one ApoB molecule attached to it. Measuring ApoB is the only way to get a true “headcount” of the particles threatening your arteries.
Think of it this way: Imagine two highways.
- Highway A has 10 massive semi-trucks carrying a heavy load of cargo.
- Highway B has 100 small cars carrying that same total weight of cargo.
If you only measure the weight, both highways look the same. But Highway B is a massive traffic jam. Because there are so many more vehicles, the chances of one of them crashing into the guardrail are much higher.
In people with “sugar issues” or obesity, the body creates many small trucks that are empty of cargo. This is why John’s LDL weight was low, but his particle headcount was likely sky-high. Leading experts now call ApoB the “most informative single circulating marker of atherogenic particle burden.” This is Tier A evidence: the unifying causal driver of heart disease.
3. Takeaway 2: The “Sticky Tape” Effect in Your Arteries
Why does the headcount matter more than the weight? It comes down to a biological process called Response-to-Retention.
Your arteries are not just smooth pipes; they are lined with a delicate layer of cells. Behind that lining is a space called the subendothelial matrix. For heart disease to start, a “bad” particle has to push its way past that lining and get stuck in that matrix.
Think of the ApoB molecule like a piece of sticky tape or Velcro. The ApoB molecule has a positive charge. The walls of your arteries contain molecules (like glycosaminoglycans) that have a negative charge. When a particle enters the artery wall, it gets “trapped” through ionic binding—essentially, the positive and negative charges snap together like magnets.
Once that particle is stuck, it doesn’t just sit there. It begins to oxidize, which “starts a fire” in your artery wall called inflammation. Your body sends in “clean-up crews” to eat the stuck particles, but they eventually turn into “foam cells,” which form the bulk of a plaque.
The rule is simple: If the particle doesn’t get stuck, the disease doesn’t start. Since every particle that causes plaque has an ApoB molecule on it, ApoB is the “glue” that makes them stick.
4. Takeaway 3: When Your Labs “Lie” to You (The Discordance Trap)
Sometimes, your standard cholesterol test and your ApoB test will tell two different stories. Scientists call this Discordance. This is the trap that caught John.
This trap is most common in people with Insulin Resistance, Metabolic Syndrome, or Obesity. In these conditions, your liver pumps out a high number of particles, but those particles are “cholesterol-depleted”—they are small, dense, and carry very little cargo. Because they are light, your LDL-C score stays low, tricking your doctor into thinking you are safe.
However, your ApoB count remains dangerously high. This is Tier C evidence for risk: your labs are essentially lying to you about the “traffic jam” in your blood. When your LDL and ApoB disagree, the 2026 medical guidelines are clear: “Treating to the higher-risk reading (ApoB) is the safer course.” If you have a large waistline or high triglycerides, you are the most likely candidate for this hidden risk.
5. Takeaway 4: It’s Not Just a Heart Problem—It’s a Brain and Body Problem
We often treat cholesterol like a “heart-only” issue, but these particles travel through every “pipe” in your body. High ApoB levels are Tier A (causal) drivers for several devastating conditions:
- Large-Artery and Small-Vessel Strokes: When these particles clog the arteries in your brain, you lose the ability to speak or move. The SPARCL trial proved that lowering these particles significantly reduces the risk of a second stroke. New data from the VESALIUS-CV (2025) trial shows that lowering them early can prevent these events before they ever happen.
- Peripheral Artery Disease (PAD): This is “clogged pipes” in the legs. It causes “claudication”—a deep, cramping pain when walking because your muscles aren’t getting enough oxygen. The Million Veteran Program found that the same genes that raise ApoB also cause PAD. Most importantly, the FOURIER trial showed a 42% reduction in major limb events (like amputations) when ApoB was aggressively lowered.
- Chronic Kidney Disease (CKD): This is Tier C People with kidney issues often have a “uremic dyslipidemia” where their ApoB is very high. While we are still mapping how much ApoB “causes” kidney failure, the SHARP trial proved that lowering these particles is a massive lifesaver for kidney patients who are at high risk for heart events.
6. Takeaway 5: The “Lp(a)” Cousin—The Heart’s Genetic Wildcard
There is a special type of ApoB particle you must know about called Lipoprotein(a), or Lp(a). This is Tier B evidence—causal, but we are still finishing the final drug trials.
Think of Lp(a) as an extra-sticky, “heavy-duty” version of an ApoB particle. You don’t get high Lp(a) from a poor diet; you inherit it from your parents. It is a genetic wildcard. Lp(a) is uniquely dangerous because it is the primary driver of Calcific Aortic Valve Stenosis—a condition where the “door” of your heart becomes stiff and covered in calcium, eventually requiring open-heart surgery.
Standard statins do not lower Lp(a). This is why some people with “perfect” scores still end up in the operating room. However, hope is on the horizon. New “gene-silencing” medicines like Pelacarsen and Olpasiran are currently in Phase 3 trials (Lp(a)HORIZON and OCEAN(a)-Outcomes). These drugs can lower this genetic risk by up to 80%.
7. Takeaway 6: The Eye-Opening Link to Diabetes and Vision
One of the most fascinating new findings is how ApoB affects your sight. People with diabetes often suffer from Diabetic Retinopathy, where the tiny blood vessels in the back of the eye become damaged.
When these vessels leak, they leave behind “hard exudates.” Doctors have discovered that these exudates are literally leaked ApoB deposits that have escaped through a damaged blood-retinal barrier. They are essentially “plaques” in your eyes.
The LENS trial (2024) provided landmark evidence that drugs which target these specific particles (like fenofibrate) can actually slow down the progression of eye disease. Managing your “particle headcount” isn’t just about avoiding a heart attack; it is about preserving your ability to see the world.
8. Takeaway 7: Could ApoB Be the Key to Preventing Dementia?
Can high cholesterol cause your brain to fail? This is the cutting edge of research, where we see a divide in the Evidence Ladder.
- Vascular Dementia (Tier B/C): This is the “clogged pipe” theory of memory loss. If the arteries in your brain become narrowed by plaque, your brain cells starve and die. This link is very strong; what is bad for the heart is almost always bad for the brain.
- Alzheimer’s Disease (Tier D): This link is “emerging.” A landmark 2026 study by Pham and colleagues found that people who are genetically prone to high ApoB have a higher risk of all-cause dementia.
While we are still in the “hypothesis-generating” stage for Alzheimer’s, the human impact is clear. Losing your memory is the ultimate loss of self. If ApoB can cross into the brain and drive inflammation, then lower ApoB levels might be one of our best tools for “successful aging.”
9. Takeaway 8: The New “Golden Rules” for Your Next Checkup
The medical world is finally catching up. The 2026 ACC/AHA and 2025 ESC/EAS guidelines now recognize that ApoB is a vital tool for understanding your true risk.
Here is your 3-step checklist for your next doctor’s visit:
- Ask for an ApoB test specifically if:
- You have Diabetes or Insulin Resistance.
- Your BMI is over 30 (Obesity).
- Your Triglycerides are over 150 mg/dL.
- You have Kidney Disease (Stage 3+).
- Check Lp(a) at least once: It’s a one-time genetic test. You need to know if you have the “extra-sticky” wildcard particle.
- Trust the Headcount: If your LDL says you are “fine” but your ApoB is “high,” trust the ApoB. Standard practice now suggests that the higher-risk reading is the one that should guide your treatment.
While universal testing for everyone isn’t the law yet, “selective testing” for anyone with metabolic risk is now the gold standard of care.
10. Conclusion: The Future of Your Health
The 20th century was the era of “cholesterol weight.” We did the best we could with the tools we had. But the 21st century is the era of the “particle headcount.”
ApoB tells a story that the standard LDL test simply cannot see. It tells us how many dangerous vehicles are actually on the road, how many are likely to get stuck in your artery walls, and how much risk you really face—not just in your heart, but in your brain, your eyes, and your limbs.
The science is converging: ApoB is the unifying driver of vascular disease. Whether it is Tier A proven science or Tier D emerging research, the signal is the same: the fewer of these particles you have, the longer your “pipes” will stay clean.
If you knew your “good” lab results were hiding a “bad” particle count, would you change your plan today? Don’t wait for a mystery event like John’s. Ask for the headcount. Ask for ApoB.
DEEP DIVE
Beyond the Coronary Artery
The Clinical Significance of Elevated Apolipoprotein B Across the Spectrum of Vascular, Metabolic, Hepatic, Renal, and Neurologic Disease
Abstract
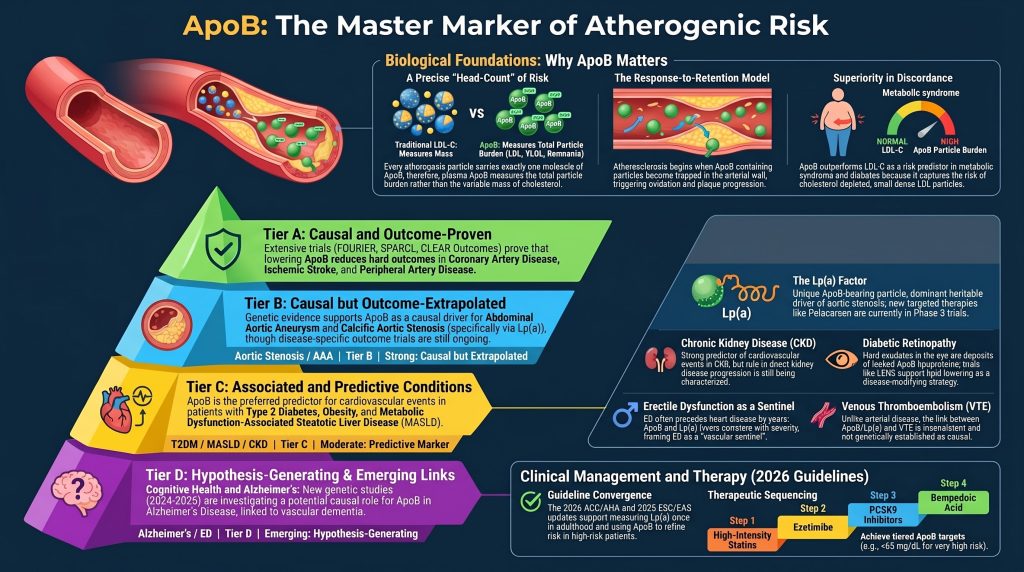
Elevated apolipoprotein B (ApoB) is the most informative single circulating marker of atherogenic particle burden and is the unifying causal driver of atherosclerotic cardiovascular disease (ASCVD). Its clinical value is greatest where ApoB and low-density lipoprotein cholesterol (LDL-C) are discordant — most often in insulin-resistant phenotypes characterized by cholesterol-depleted small dense LDL and triglyceride-rich remnants. Beyond classic ASCVD, the relationship between elevated ApoB and disease ranges from causal-and-outcome-proven (ischemic stroke, peripheral artery disease), to causal-but-outcome-extrapolated (abdominal aortic aneurysm, calcific aortic stenosis via lipoprotein(a)), to associated and predictive (metabolic dysfunction-associated steatotic liver disease, chronic kidney disease, diabetic retinopathy), to hypothesis-generating (Alzheimer disease, erectile dysfunction, venous thromboembolism, cancer outcomes). This review structures the evidence into a transparent ladder so that the strength of inference matches the strength of the underlying data, summarizes contemporary and forthcoming therapeutics by outcome status, and aligns recommendations with the 2026 ACC/AHA dyslipidemia guideline, the 2025 ESC/EAS focused update, the 2021 Canadian Cardiovascular Society guideline, and recent National Lipid Association consensus.
An Evidence Ladder for ApoB and Disease
To prevent overgeneralization, every disease state in this review is graded on a four-tier evidence ladder. The label drives the strength of the recommendation in the section that follows.
- Tier A — Causal and outcome-proven: Mendelian randomization (MR) supports causality AND randomized controlled trials (RCTs) that lower ApoB-containing particles reduce hard outcomes in this disease, with prespecified or robust subgroup analyses.
- Tier B — Causal but outcome-extrapolated: MR or strong genetic evidence supports causality, BUT outcome-reduction data are extrapolated from related ASCVD endpoints rather than disease-specific RCTs.
- Tier C — Associated and predictive: Robust observational and mechanistic data link ApoB to the disease and ApoB predicts events, BUT causality is not established by MR or treatment evidence is mixed.
- Tier D — Hypothesis-generating: Mechanistic plausibility plus limited observational signals; no convincing causal or interventional evidence.
Where a disease has heterogeneous evidence across subtypes (e.g., vascular cognitive impairment vs. Alzheimer disease; ischemic vs. hemorrhagic stroke; CKD events vs. progression), each subtype is graded separately rather than averaged. Where a disease sits between two tiers because evidence is partial — for example, hypertensive vascular disease (synergistic with atherosclerosis but limited disease-specific RCT data) — a dual designation such as “B/C” is used and explained in the relevant section. The intent is descriptive transparency, not pseudo-precise scoring.
Part I — Biological Foundations
ApoB Counts Atherogenic Particles
Each LDL, intermediate-density lipoprotein (IDL), very-low-density lipoprotein (VLDL), chylomicron remnant, and lipoprotein(a) [Lp(a)] particle carries exactly one molecule of apolipoprotein B — apoB-100 on hepatically secreted particles, apoB-48 on intestinally secreted ones [1, 2]. Plasma ApoB is therefore a head-count of atherogenic particles, whereas LDL-C is a mass measurement that depends on a variable cholesterol-per-particle stoichiometry [3, 4]. When the average cholesterol cargo per particle falls — as happens in insulin-resistant states with cholesterol-depleted small dense LDL — the same plasma cholesterol mass corresponds to a larger number of particles, and ApoB rises out of proportion to LDL-C. This is the source of clinically meaningful ApoB / LDL-C discordance and the principal reason ApoB outperforms LDL-C in metabolic syndrome, type 2 diabetes, MASLD, and obesity [5, 6, 7].
The Response-to-Retention Mechanism in the Arterial Wall
Atherosclerosis begins when ApoB-containing particles cross the endothelium and become trapped in the subendothelial extracellular matrix through ionic binding between positively charged residues on apoB-100 and negatively charged glycosaminoglycans on biglycan and decorin [8, 9]. Retained particles are oxidized, drive macrophage foam-cell formation, activate the NLRP3 inflammasome, and propagate plaque progression [10]. This response-to-retention model is a property of arterial atherosclerosis and applies to coronary, carotid, cerebral, peripheral, renal, and aortic arteries. Extension of the same mechanism to non-arterial vascular beds — hepatic sinusoids, glomerular mesangium, retinal capillaries, cavernosal microvessels — is biologically plausible but evidentiarily weaker, and is treated as such in the disease-by-disease sections that follow.
Mendelian Randomization: From Association Toward Causation
Genetically lower ApoB confers lifelong protection against coronary heart disease and several extra-coronary outcomes. Multivariable MR analyses by Richardson and colleagues (PLoS Medicine, 2020) and Marston and colleagues (JAMA Cardiology, 2022) show that when ApoB is held constant, the residual associations of LDL-C and triglycerides with myocardial infarction substantially attenuate — supporting the interpretation that ApoB-containing particle burden is the dominant causal lipid signal for ASCVD, with cholesterol and triglyceride content acting as cargo rather than as independent risk factors [11, 12]. ApoB is necessary but not always sufficient: remnant cholesterol, Lp(a), oxidized phospholipids, endothelial biology, and systemic inflammation contribute residual risk beyond ApoB-particle counts. With those caveats noted, the convergence of MR, cumulative-exposure modeling, and randomized trials of mechanistically distinct ApoB-lowering drugs achieving similar per-mg/dL benefit constitutes strong — though not absolute — evidence of causality, with the well-known MR assumptions (pleiotropy, canalization, equivalence of lifelong genetic exposure to pharmacologic exposure) acknowledged as limitations [13].
Part II — Tier A: Causal and Outcome-Proven Disease
Coronary Artery Disease and Myocardial Infarction (the ApoB vs LDL-C Discriminator)
Treated here as a discriminator analysis, since the question is what ApoB adds beyond LDL-C and non-HDL-C, not whether atherosclerotic CAD is ApoB-driven (it is). The 2011 Sniderman meta-analysis (n = 233,455) reported standardized relative risks of 1.43 for ApoB, 1.34 for non-HDL-C, and 1.25 for LDL-C [14]. The differences between ApoB and non-HDL-C are clinically modest in concordant populations, and both metrics remain reasonable secondary targets endorsed by current guidelines. The 2022 Marston UK Biobank analysis (n = 389,529) demonstrated that ApoB substantially attenuated the risk associated with LDL-C and triglycerides; once ApoB was in the model, LDL-C and triglycerides contributed little additional information [12]. Behbodikhah and colleagues (2021) and Glavinovic and colleagues (2022) formalized ApoB as the dominant — though not exclusive — unifying causal particle [4, 5]. When ApoB and LDL-C disagree, treating to the higher-risk reading is the safer course; when they concord, either metric is clinically defensible.
Ischemic Stroke (Large-Artery and Small-Vessel)
MR studies including MEGASTROKE (Hindy and colleagues, 2018) and the wide-angled MR by Allara and colleagues (2019) show that genetically elevated LDL-C and ApoB causally increase risk of large-artery atherosclerotic ischemic stroke and small-vessel stroke; effects on cardioembolic stroke are null [15, 16]. SPARCL demonstrated that high-intensity atorvastatin reduces recurrent stroke after stroke or TIA [17]. FOURIER (evolocumab) and ODYSSEY OUTCOMES (alirocumab) reduced ischemic stroke proportionally to ApoB lowering, without increasing hemorrhagic stroke at LDL-C as low as <30 mg/dL [18, 19].
The hemorrhagic-stroke literature is more nuanced and the optimal lower threshold for LDL-C and ApoB remains debated. Sun and colleagues reported a modest positive association between very low LDL-C and intracerebral hemorrhage in Chinese adults [20]. Absolute event rates at LDL-C <40 mg/dL are small, and FOURIER and ODYSSEY did not show a hemorrhagic-stroke signal. On balance the trial evidence supports a net cerebrovascular benefit of lowering in high-risk ASCVD populations, but caution remains warranted in poorly controlled hypertensives, in some East Asian cohorts, and at very low achieved LDL-C values where the absolute benefit-to-harm ratio is less well characterized.
The 2025 VESALIUS-CV trial extended this evidence by showing that adding evolocumab to optimized lipid therapy in high-cardiovascular-risk patients without prior myocardial infarction or stroke reduced atherosclerotic events, supporting the lower-for-longer paradigm into earlier disease stages [21].
Peripheral Artery Disease
Klarin and colleagues (Nature Medicine, 2019) used the Million Veteran Program to identify and replicate genetic determinants of PAD that overlap with LDL-C–raising loci, supporting causality of ApoB-containing particles [22]. The FOURIER PAD subgroup (Bonaca and colleagues, 2018) demonstrated a 42% reduction in major adverse limb events at the lowest achieved LDL-C [23]. CLEAR Outcomes (Nissen and colleagues, 2023) showed bempedoic acid reduces a composite cardiovascular endpoint that included limb events in statin-intolerant patients [24]. ApoB outperforms LDL-C in diabetic PAD specifically because of the small-dense-LDL and remnant phenotype [6].
Part III — Tier B: Causal but Outcome-Extrapolated Disease
Abdominal Aortic Aneurysm
Harrison and colleagues (JAMA Cardiology, 2018) and Allara and colleagues (2019) used MR to show that LDL-C and ApoB-raising variants causally raise AAA risk [16, 25]. Statin meta-analyses suggest slowed aneurysm growth, but disease-specific RCTs powered for hard outcomes are limited; the reduction in aortic events in trials such as FOURIER reinforces the causal direction [18, 25].
Calcific Aortic Valve Stenosis (Lp(a) Specifically)
Calcific aortic stenosis is the disease most uniquely driven by Lp(a) — an ApoB-bearing particle. Thanassoulis and colleagues (NEJM, 2013) used MR with LPA variants (rs10455872) to demonstrate that Lp(a) causally raises CAVS risk independent of LDL-C [26]. Subsequent work by Kamstrup, Nordestgaard, and Tsimikas confirmed Lp(a) as a dominant heritable driver of CAVS, with the relevant pathobiology involving Lp(a)-borne oxidized phospholipids initiating valvular inflammation and calcification [27, 28]. Statins do not slow CAVS progression (ASTRONOMER, SEAS, SALTIRE) — consistent with Lp(a) being the dominant target — and Lp(a)-lowering therapies are now in advanced development.
Lp(a)-Targeted Therapies — Current Status
To prevent inflated expectations, the developmental status of each agent should be stated precisely:
- Pelacarsen (TQJ230): antisense oligonucleotide. The 2020 NEJM paper by Tsimikas and colleagues was a phase 2 dose-ranging study demonstrating up to 80% Lp(a) reduction [29]. The phase 3 cardiovascular outcomes trial Lp(a)HORIZON is ongoing, with completion expected in 2026–2027 [30].
- Olpasiran: small-interfering RNA. The 2022 NEJM OCEAN(a)-DOSE paper was a phase 2 dose-ranging study; the phase 3 outcomes trial OCEAN(a)-Outcomes is ongoing [31, 32].
- Lepodisiran: siRNA in advanced development; the phase 3 outcomes trial ACCLAIM-Lp(a) is now enrolling [33].
- Muvalaplin: first-in-class oral small-molecule inhibitor of Lp(a) assembly with phase 3 outcomes development announced [34].
No completed phase 3 outcomes trial of any Lp(a)-specific therapy has yet been reported. Outcome-reduction claims are therefore extrapolated from per-particle ApoB biology, MR, and the established vascular toxicity of Lp(a).
Part IV — Tier C: Associated and Predictive Conditions
Type 2 Diabetes Mellitus
ApoB is consistently elevated in T2DM, and discordance with LDL-C is a defining feature of diabetic dyslipidemia (high triglycerides, low HDL-C, normal-to-modestly-elevated LDL-C, elevated non-HDL-C and ApoB) [6, 35]. ApoB outperforms LDL-C as a predictor of cardiovascular events in T2DM, and the 2021 Canadian Cardiovascular Society guideline preferentially recommends ApoB or non-HDL-C in diabetes and hypertriglyceridemia [36]. The 2026 ACC/AHA guideline supports selective use of ApoB to refine residual risk in cardiometabolic-kidney syndrome, T2DM, hypertriglyceridemia, and established CVD [37]. Whether ApoB is itself causal for incident T2DM remains debated. A multivariable Mendelian randomization analysis by Richardson and colleagues (Lancet Healthy Longevity, 2021) found that ApoB behaved differently in univariable vs. multivariable models and that the multivariable signal pointed toward increased T2DM risk — consistent with the mechanistic proposal that β-cell cholesterol exposure (mediated by ABCA1) impairs insulin secretion [38, 39] — but the directionality is complicated by the well-known modest increase in T2DM incidence with statin therapy. The dominant clinical message in T2DM is therefore predictive and treatment-targeted rather than incidence-causal. CARDS, HPS-DIABETES, and the diabetes subgroup of REDUCE-IT (icosapent ethyl 4 g/day in statin-treated patients with elevated triglycerides) show meaningful event reduction [40, 41].
Insulin Resistance and Metabolic Syndrome
In insulin resistance, hepatic VLDL secretion increases, plasma residence time of ApoB-containing particles lengthens, and CETP-mediated lipid exchange combined with hepatic-lipase trimming generates small-dense LDL. The net result is the canonical discordance: more particles carrying less cholesterol each. Cromwell and colleagues (Framingham Offspring) and Mora (Women’s Health Study) showed that LDL-particle number tracks more closely with events than LDL-C in this population [42, 43]. Lifestyle interventions, GLP-1 receptor agonists, and SGLT2 inhibitors all lower ApoB modestly through weight, triglyceride, and remnant effects [44].
Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD/MASH)
MASLD shares an upstream driver with atherogenic dyslipidemia: hepatic de novo lipogenesis and VLDL overproduction. Patients with MASLD typically have elevated ApoB, elevated remnant cholesterol, and small-dense LDL — often with apparently normal LDL-C [45, 46]. The cardiovascular implications matter clinically: cardiovascular disease is the leading cause of death in MASLD, and ApoB outperforms LDL-C as a risk discriminator in this population [46]. Statins are safe and recommended in MASLD/MASH per AASLD and EASL guidance [47]. Resmetirom, a thyroid-hormone receptor-β agonist, was approved by the FDA in March 2024 for non-cirrhotic MASH with moderate-to-advanced fibrosis on the basis of the MAESTRO-NASH trial; it lowers ApoB and LDL-C while improving histology, although cardiovascular outcomes data are not yet available [48].
On causality: PNPLA3 (I148M) and TM6SF2 (E167K) variants reduce hepatic VLDL secretion and lower ApoB while paradoxically increasing intrahepatic lipid accumulation and MASLD progression — illustrating that hepatic ApoB export is partially protective against intrahepatic lipid burden but increases circulating atherogenic load [49, 50]. The relationship between ApoB and MASLD is therefore best described as bidirectional and metabolically intertwined, rather than as ApoB causing MASLD in the same sense that ApoB causes atherosclerosis.
Chronic Kidney Disease
CKD produces a uremic dyslipidemia characterized by elevated triglycerides, reduced HDL-C, often low-to-normal LDL-C, and elevated ApoB and Lp(a) due to impaired remnant clearance and apo(a) accumulation [51]. SHARP (simvastatin/ezetimibe in CKD) reduced major atherosclerotic events by 17%; benefit attenuated in dialysis patients (4D, AURORA were null), reflecting the shift from atherosclerotic to non-atherosclerotic cardiovascular death at end-stage disease [52, 53]. ApoB predicts cardiovascular events in CKD better than LDL-C in post-hoc analyses of these trials. The Lanktree and colleagues 2018 American Journal of Kidney Diseases MR analysis examined the relationship between HDL-C, LDL-C, triglycerides, and CKD risk and found mixed signals, supporting that lipid effects on CKD progression itself are smaller than effects on CKD-associated cardiovascular events [54]. The mechanistic literature on glomerular mesangial foam cell formation and lipid nephrotoxicity is biologically coherent but does not yet meet a causal threshold for CKD progression.
Hypertensive Vascular Disease
ApoB and hypertension act independently and synergistically on atherosclerosis. Hypertension increases endothelial permeability, while ApoB provides the substrate for retention. Both contribute to arterial stiffening, left ventricular hypertrophy, and end-organ damage. SCORE2 and the Pooled Cohort Equations integrate both BP and lipid measurements; whether ApoB adds prognostic discrimination beyond non-HDL-C in SCORE2 has been formally evaluated. A 2025 analysis by Wong, Takeuchi, Thao, Nicholls, Chew, and Peter in the European Journal of Preventive Cardiology found that adding ApoB to SCORE2 did not materially improve discrimination, calibration, or net reclassification, although ApoB cutoffs combined with SCORE2 thresholds refined classification at the margins [55]. Current evidence therefore does not support replacing standard SCORE2 inputs with ApoB; ApoB is best used as a complementary residual-risk metric.
Obesity and Bariatric/Pharmacologic Weight Loss
Visceral adiposity drives hepatic VLDL overproduction and elevates ApoB. Weight-loss interventions reduce ApoB: bariatric surgery in meta-analyses, GLP-1 receptor agonists (with the SELECT trial demonstrating cardiovascular event reduction with semaglutide in obesity without diabetes, alongside meaningful ApoB and lipid effects), and to a lesser extent SGLT2 inhibitors, all lower ApoB substantially in parallel with adiposity reduction [56]. Obese patients commonly have apparently normal LDL-C with markedly elevated ApoB; Welsh and colleagues (Circulation, 2021) showed in UK Biobank that ApoB outperforms LDL-C as a predictor across BMI strata [57]. The lean-mass-hyper-responder phenotype — lean, insulin-sensitive individuals on ketogenic diets who develop very high LDL-C and ApoB — has prompted observational debate (KETO-CTA), but the published cohort is uniformly at extreme ApoB and lacks a low-ApoB control, limiting inference. The dominant body of MR and RCT evidence on ApoB causality is not overturned by a single observational study at restricted ApoB range.
Familial Hypercholesterolemia
Heterozygous familial hypercholesterolemia (HeFH; prevalence ~1 in 250) and homozygous FH (HoFH; ~1 in 300,000) are monogenic disorders of LDLR, APOB (familial defective ApoB), or PCSK9 gain-of-function — directly elevating ApoB. Lifetime ApoB exposure is the mechanism of premature ASCVD; HoFH patients can present with myocardial infarction in the first or second decade. Therapy is ApoB-directed: high-intensity statins, ezetimibe, PCSK9 monoclonal antibodies (alirocumab, evolocumab) for HeFH and HoFH (residual LDLR function), evinacumab (ANGPTL3 monoclonal; ELIPSE-HoFH, NEJM 2020), lomitapide, and LDL apheresis where needed [58, 59]. FH is among the strongest natural experiments supporting ApoB causality.
Hypertriglyceridemia, Mixed Dyslipidemia, and Remnant Cholesterol
ApoB captures the atherogenic burden in hypertriglyceridemia better than any other single test because it counts each VLDL, IDL, and remnant particle. Remnant cholesterol — calculated or measured — is causally atherogenic per MR analyses by Varbo, Nordestgaard, and colleagues [60, 61]. REDUCE-IT showed that icosapent ethyl 4 g/day reduces events by 25% in statin-treated patients with triglycerides 135–499 mg/dL [40], although recent expert consensus has tempered the strength of recommendation given unresolved questions about the comparator (mineral oil). PROMINENT showed that pemafibrate lowered triglycerides and remnant cholesterol without lowering ApoB and did not reduce cardiovascular events — in fact slightly increasing ApoB — providing a powerful natural experiment in support of the principle that ApoB-particle reduction, not triglyceride reduction per se, is the therapeutic objective [62]. Investigational agents olezarsen and plozasiran (APOC3-directed) and zodasiran (ANGPTL3 siRNA) lower ApoB-containing particle count and triglycerides; cardiovascular outcomes trials are pending. Olezarsen received FDA approval in December 2024 for familial chylomicronemia syndrome to reduce pancreatitis risk — a rare phenotype-specific indication that should not be conflated with proven ASCVD event reduction [63, 64]. The unifying conclusion: remnant-rich, ApoB-containing particles are atherogenic and constitute a real residual-risk target, but not every mixed-dyslipidemia phenotype yet has dedicated ApoB-lowering outcome trials.
Diabetic Retinopathy
Beyond glycemic and BP control, dyslipidemia — and particularly ApoB-containing remnant lipoproteins — predicts diabetic retinopathy severity, diabetic macular edema, and progression [65]. The FIELD trial (fenofibrate, 2007) and the ACCORD-Eye fenofibrate-plus-simvastatin substudy showed approximately 40% reductions in DR progression — substantially independent of glycemic effect — attributed to remnant lipoprotein lowering and direct PPAR-α anti-inflammatory effects in retinal endothelium [66, 67]. The 2024 LENS trial provides updated randomized evidence in early DR, supporting fenofibrate as a disease-modifying therapy in this microvascular complication [68]. Hard exudates in DR are histologically deposits of ApoB-containing lipoproteins extravasated through a damaged blood-retinal barrier [69]. The mechanistic and clinical evidence is strong; whether ApoB itself is causal versus a marker of remnant burden remains debated, and fenofibrate’s benefit may operate through pleiotropic pathways.
Pregnancy-Related Complications
Pregnancy is a physiologically dyslipidemic state. Pre-pregnancy and early-pregnancy ApoB elevations associate with later preeclampsia, gestational diabetes, and preterm birth in cohort studies [70]. The mechanistic links involve endothelial dysfunction (preeclampsia) and pre-existing insulin resistance (gestational diabetes). The FDA in 2021 removed the blanket strongest warning against statin use in pregnancy, but this is not a general endorsement; current evidence on pravastatin for preeclampsia prevention from trials including StAmP and INOVASIA is mixed, with meta-analytic uncertainty [71, 72]. Statins should not be initiated routinely in pregnancy outside trial settings or after individualized maternal-fetal medicine consultation.
Vascular Cognitive Impairment
Vascular cognitive impairment (VCI) shares its pathophysiology with stroke and small-vessel disease; ApoB-driven cerebral atherosclerosis and lipohyalinosis cause the cumulative white-matter-hyperintensity burden, lacunes, and microbleeds that manifest as vascular cognitive decline [73, 74]. The vascular dementia case for ApoB is correspondingly strong: it inherits the causal evidence from ischemic stroke and small-vessel disease.
Alzheimer Disease (Emerging)
For Alzheimer disease (AD) the picture is more uncertain and more confounded. APOE ε4 is the dominant genetic risk factor and participates in lipoprotein metabolism but is distinct from ApoB. A 2026 multivariable Mendelian randomization study by Pham, Mulugeta, Lumsden, and Hyppönen (GeroScience, April 2026) reported that ApoB was associated with higher all-cause dementia risk in multivariable MR, although the signal was sensitive to model specification [75]. A 2024 Communications Biology analysis by Adams, Martin and colleagues separately linked genetically predicted ApoB (but not LDL-C) to Alzheimer risk, lending support to a Tier D hypothesis-generating role [90]. Iwagami and colleagues (Lancet Healthy Longevity, 2021) showed in 1.8 million people that midlife elevated total cholesterol associates with late-life dementia [76]. Statin meta-analyses suggest reduced dementia incidence with midlife use, but trial evidence (PROSPER, HPS) is mixed and underpowered [77]. Recent observational data also link elevated Lp(a) to brain infarcts and dementia [78]. The Alzheimer case for ApoB therefore remains emerging — supported by mechanistic plausibility and a small, mixed MR base, but not at the strength of the vascular cognitive impairment argument.
Part V — Tier D: Hypothesis-Generating Conditions
Erectile Dysfunction
Erectile dysfunction often precedes coronary disease by 3–5 years because the cavernosal artery is small (1–2 mm) and shows endothelial dysfunction earlier [79]. ApoB and Lp(a) correlate with ED severity in cross-sectional studies, and statin therapy modestly improves erectile function in meta-analyses, plausibly via endothelial recovery [80]. The literature is largely observational; ED is best framed as a vascular sentinel, not a separately ApoB-causal disease.
Retinal Vein Occlusion
Retinal vein occlusion has been associated with elevated ApoB and Lp(a) in observational studies; mechanistically it shares atherothrombotic features with arterial vascular disease [81]. Causality is not established.
Venous Thromboembolism
Historically considered distinct from atherogenic risk. The Lp(a)–VTE relationship is biologically plausible — Lp(a) is antifibrinolytic (through apo(a) homology with plasminogen) and carries oxidized phospholipids — but the published evidence is inconsistent. Recent European Heart Journal analyses describe the Lp(a)–VTE relationship as not genetically established, in contrast to the strong arterial and valvular signals; one MR study found no statistically significant causal effect of ApoB, LDL-C, HDL-C, triglycerides, or apoA1 on DVT [82, 83]. Recent work also suggests sex- and hormone-dependent heterogeneity rather than a generalizable causal effect. JUPITER post-hoc analyses suggest modest VTE benefit with rosuvastatin [84]. The most defensible conclusion is that the relationship is inconsistent and the signal, if real, is modest.
Cancer Outcomes
Evidence is heterogeneous and largely associative. Some MR work suggests low LDL-C/ApoB associates with higher risk of certain cancers — most likely reflecting reverse causation from preclinical malignancy lowering circulating cholesterol — while observational cohort data link elevated ApoB with obesity-related cancers. Causality is not established and low ApoB should not be construed as a cancer-prevention strategy [85].
Part VI — ApoB-Lowering Therapies, by Evidence Status
Lumping all ApoB-lowering agents together overstates the certainty of benefit for newer agents. The following three-tier organization mirrors the evidence ladder used for diseases.
Outcome-Proven for ASCVD Risk Reduction
- Statins (rosuvastatin, atorvastatin, others) — large body of RCT evidence across primary and secondary prevention.
- Ezetimibe — IMPROVE-IT demonstrated added benefit on top of statin therapy.
- PCSK9 monoclonal antibodies (alirocumab, evolocumab) — FOURIER, ODYSSEY OUTCOMES, and the 2024–2025 VESALIUS-CV trial extending benefit to high-risk patients without prior MI/stroke [18, 19, 21].
- Bempedoic acid — CLEAR Outcomes (2023) in statin-intolerant patients [24].
Outcome Benefit in Specific Phenotypes
- Icosapent ethyl — REDUCE-IT (statin-treated patients with persistent hypertriglyceridemia, primarily for cardiovascular events) [40]. Note that recent expert consensus has reduced its strength of recommendation in some guidelines because of unresolved questions about the placebo (mineral oil).
- Fenofibrate — FIELD, ACCORD-Eye, and LENS for diabetic retinopathy progression; not generally indicated for ASCVD event reduction [66, 67, 68].
Investigational or Niche Therapies
- Inclisiran — siRNA-based PCSK9 inhibitor; dramatic and durable LDL-C/ApoB lowering. The cardiovascular-outcomes trial ORION-4 is ongoing and the 2026 ACC/AHA guideline notes that outcomes data are still pending [37, 86]. Notwithstanding, twice-yearly dosing has given inclisiran a meaningful niche role for adherence-challenged patients, and the 2025 ESC/EAS focused update gives a stronger Class I/IIa recommendation depending on risk category [87].
- Lp(a)-targeted therapies (pelacarsen, olpasiran, lepodisiran, muvalaplin) — phase 3 outcomes trials Lp(a)HORIZON, OCEAN(a)-Outcomes, and ACCLAIM-Lp(a) are ongoing [30, 32, 33].
- APOC3-directed agents (olezarsen, plozasiran) — olezarsen is FDA-approved for familial chylomicronemia syndrome (pancreatitis prevention); ASCVD outcomes are not yet established [63, 64].
- ANGPTL3-directed agents (evinacumab approved for HoFH; zodasiran in development) — outcomes for non-FH ASCVD are not yet established [58].
Part VII — The Contemporary Guideline Landscape
As of 2025–2026 the major guidelines have evolved meaningfully from the 2018 ACC/AHA cholesterol guideline framework:
- The 2026 ACC/AHA dyslipidemia guideline (replacing the 2018 cholesterol guideline) reintroduces LDL-C and non-HDL-C treatment goals, recommends Lp(a) measurement at least once in adulthood, and supports selective ApoB testing to assess residual risk — particularly in cardiometabolic-kidney syndrome, T2DM, hypertriglyceridemia, and known CVD [37].
- The 2025 ESC/EAS focused update to the 2019 dyslipidemia guideline incorporates evidence published through March 2025 and continues to support ApoB targets in high- and very-high-risk patients [87].
- The 2021 Canadian Cardiovascular Society guideline preferentially recommends ApoB or non-HDL-C, particularly when triglycerides exceed 1.5 mmol/L or in cardiometabolic disease [36].
- Recent National Lipid Association consensus statements broaden the practical role of ApoB testing in residual-risk assessment [88].
The synthesis: there is convergence across societies that ApoB is clinically valuable, particularly for residual risk and for discordant LDL-C/ApoB phenotypes, but no major society currently recommends ApoB as the universal first-line lipid screen for every adult.
Part VIII — Practical Recommendations from the Guidelines
Selective ApoB Testing
Measure ApoB at least once in any adult with type 2 diabetes, metabolic syndrome, MASLD, obesity (BMI ≥30), CKD stages 3 and higher, fasting triglycerides ≥150 mg/dL, known or suspected familial hypercholesterolemia, family history of premature ASCVD, or LDL-C in the 70–190 mg/dL range where treatment intensity is uncertain. This aligns with ESC/EAS, CCS, and the selective use endorsed by 2026 ACC/AHA. Universal ApoB screening of all adults is not currently a guideline-endorsed practice.
Increasingly Recommended Lp(a) Measurement
Measure Lp(a) at least once in every adult where guideline-aligned practice permits. The recommendation is endorsed by the 2025 ESC/EAS focused update, the 2026 ACC/AHA guideline, and prior 2019 ESC/EAS guidance, and is increasingly — though not yet universally — implemented across health systems. Lp(a) is critical in calcific aortic stenosis evaluation, in premature MI, and in family history of premature ASCVD; it has prognostic value across primary and secondary prevention.
Treatment Targets
Use LDL-C as the primary treatment target consistent with 2026 ACC/AHA, with ApoB as a complementary residual-risk metric — particularly when LDL-C and ApoB are discordant. ESC/EAS-aligned practice may use ApoB targets directly: very-high-risk <65 mg/dL, high-risk <80 mg/dL, moderate-risk <100 mg/dL. When the two metrics disagree, treat to the higher-risk reading.
Therapy Sequencing
- First-line: high-intensity statin (rosuvastatin 20–40 mg or atorvastatin 40–80 mg).
- Add ezetimibe 10 mg for additive ApoB lowering and outcome benefit.
- Add a PCSK9 monoclonal antibody (alirocumab or evolocumab) in very-high-risk patients not at goal.
- Use bempedoic acid in statin-intolerant patients per CLEAR Outcomes.
- Use icosapent ethyl in statin-treated patients with persistent hypertriglyceridemia and ASCVD per REDUCE-IT, with awareness of recent guideline-strength caveats.
- For Lp(a)-driven disease, consider trial enrollment in Lp(a)HORIZON, OCEAN(a)-Outcomes, ACCLAIM-Lp(a), or related programs.
- Inclisiran is reasonable for selected statin-eligible patients needing further LDL-C/ApoB reduction; outcomes data from ORION-4 are pending.
Residual Inflammatory Risk
In secondary-prevention patients at low ApoB (e.g., <60 mg/dL on therapy) with persistent hsCRP >2 mg/L and recurrent events, consider colchicine 0.5 mg daily per LoDoCo2 (FDA-approved 2023 for ASCVD risk reduction), rather than further ApoB lowering [89].
Caveats and Limitations
Mendelian randomization rests on assumptions — pleiotropy, canalization, and the equivalence of lifelong genetic exposure to drug exposure — that are imperfect. The convergence of MR with multiple drug-class RCTs (statins, ezetimibe, PCSK9 monoclonal antibodies, bempedoic acid) targeting ApoB through different mechanisms is the strongest practically attainable evidence for causality in adult populations, but it is not equivalent to a lifelong randomized trial and should not be presented as logically irrefutable.
Hemorrhagic stroke at very low LDL-C/ApoB: data are mixed; absolute risk at LDL-C <40 mg/dL is small, and net cerebrovascular benefit in trials remains favorable, but caution remains in poorly controlled hypertensives and in some East Asian cohorts.
The lean-mass-hyper-responder / KETO-CTA discussion is observational and limited by range-restriction in a uniformly extreme-ApoB cohort lacking low-ApoB controls. The dominant body of MR plus RCT evidence for ApoB causality is not overturned by an observational study of 100 individuals at restricted ApoB range.
Cancer–ApoB associations most likely reflect reverse causation and confounding.
Pregnancy data are largely observational; statins should not be initiated routinely in pregnancy outside trial settings or specialist consultation.
Assay standardization: ApoB measurement is now well-standardized using immunoturbidimetric or immunonephelometric methods calibrated to the WHO/IFCC SP3-07 reference standard. Older assays varied and historical comparisons should be interpreted accordingly.
Summary Table: ApoB Across Disease States
| Disease State | Evidence Tier | Causal vs. Associative | Mechanism | Lowering ApoB Reduces Risk? |
| CAD / MI (vs LDL-C as discriminator) | A | Causal | Subendothelial particle retention; foam-cell formation | Yes — extensive RCT evidence |
| Ischemic stroke (large-artery, small-vessel) | A | Causal | Cerebral arterial atherosclerosis; same as CAD | Yes — SPARCL, FOURIER, ODYSSEY |
| Peripheral artery disease | A | Causal | Lower-extremity arterial atherosclerosis | Yes — FOURIER limb subgroup, CLEAR |
| Hemorrhagic stroke | C | Equivocal/possibly inverse | Vessel fragility at very low LDL-C in some populations | Net cerebrovascular benefit favors lowering |
| Abdominal aortic aneurysm | B | Causal (MR) | Medial degeneration with atherosclerosis | Likely — extrapolated/limited RCT |
| Calcific aortic stenosis (Lp(a)-driven) | B | Causal (Lp(a)-MR) | Lp(a)/OxPL-driven valvular inflammation and calcification | Lp(a)-targeted phase 3 trials ongoing |
| T2DM (CV risk discrimination) | C | Predictive | Small-dense LDL, remnant accumulation | Yes for CV events; statins/PCSK9i, REDUCE-IT |
| T2DM (incidence) | C | Possibly contributory | β-cell cholesterol exposure (debated) | Unclear; not the dominant clinical message |
| Insulin resistance / metabolic syndrome | C | Predictive/contributory | VLDL overproduction, remnants, sdLDL | Yes — lifestyle, GLP-1, statins |
| MASLD / MASH | C | Bidirectional/contributory | Hepatic VLDL overproduction; cardiovascular co-morbidity | Indirect; statins safe; resmetirom approved |
| CKD (CV events) | C | Predictive | Uremic dyslipidemia; remnants/Lp(a) | Yes — SHARP for non-dialysis CKD |
| CKD (progression) | D | Hypothesis-generating | Mesangial foam-cell formation | Mixed evidence |
| Hypertensive vascular disease | B/C | Synergistic contributor | Increased permeability + ApoB substrate | Yes — additive in trials |
| Obesity-related cardiometabolic disease | C | Contributory | Visceral adiposity → hepatic ApoB output | Yes — bariatric, GLP-1 |
| Familial hypercholesterolemia | A | Causal (monogenic) | Lifelong elevated ApoB exposure | Yes — statins, PCSK9i, evinacumab in HoFH |
| Hypertriglyceridemia / mixed dyslipidemia (remnant-driven) | A/B | Causal (remnant particles) | Remnant retention; sdLDL; PROMINENT shows TG-lowering without ApoB-lowering is inert | Yes for ApoB-lowering arms (statins, ezetimibe, PCSK9i); icosapent ethyl with caveats |
| Severe HTG / familial chylomicronemia | B/C | Contributory (pancreatitis) | Chylomicron-driven; apoB-48 burden | Olezarsen FDA-approved for FCS |
| Diabetic retinopathy / DME | C | Contributory | Hard exudate deposition; PPAR-α effects | Yes — fenofibrate (FIELD, ACCORD-Eye, LENS) |
| Vascular dementia / cognitive impairment | B/C | Causal-likely (vascular) | Cerebral atherosclerosis; small-vessel disease | Likely; midlife statin associations |
| Alzheimer disease | D | Emerging | BBB transcytosis; possible amyloid-clearance link | Unclear; trial evidence underpowered |
| Erectile dysfunction | D | Predictive (vascular sentinel) | Cavernosal endothelial dysfunction | Modest — statin meta-analyses |
| Retinal vein occlusion | D | Associated | Atherothrombotic mechanisms | Likely contributory |
| Pregnancy (preeclampsia, GDM) | C/D | Predictive/contributory | Endothelial dysfunction; pre-existing IR | Mixed (pravastatin trials inconclusive) |
| Venous thromboembolism | D | Inconsistent; not genetically established | Antifibrinolysis; oxidized phospholipids (Lp(a)) | Modest at best; statin meta-analyses mixed |
| Cancer outcomes | D | Inconclusive | Pleiotropic; possible reverse causation | Not a cancer-prevention strategy |
Tier legend: A — Causal and outcome-proven; B — Causal but outcome-extrapolated; C — Associated and predictive; D — Hypothesis-generating.
References
References are formatted in IEEE numerical style. All entries refer to peer-reviewed primary literature, society guidelines, or authoritative consensus statements verifiable in PubMed/CrossRef.
- Ference BA, Ginsberg HN, Graham I, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2017;38(32):2459-2472. doi:10.1093/eurheartj/ehx144
- Borén J, Chapman MJ, Krauss RM, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2020;41(24):2313-2330. doi:10.1093/eurheartj/ehz962
- Sniderman AD, Thanassoulis G, Glavinovic T, et al. Apolipoprotein B Particles and Cardiovascular Disease: A Narrative Review. JAMA Cardiol. 2019;4(12):1287-1295. doi:10.1001/jamacardio.2019.3780
- Behbodikhah J, Ahmed S, Elyasi A, et al. Apolipoprotein B and Cardiovascular Disease: Biomarker and Potential Therapeutic Target. Metabolites. 2021;11(10):690. Published 2021 Oct 8. doi:10.3390/metabo11100690
- Glavinovic T, Thanassoulis G, de Graaf J, Couture P, Hegele RA, Sniderman AD. Physiological Bases for the Superiority of Apolipoprotein B Over Low-Density Lipoprotein Cholesterol and Non-High-Density Lipoprotein Cholesterol as a Marker of Cardiovascular Risk. J Am Heart Assoc. 2022;11(20):e025858. doi:10.1161/JAHA.122.025858
- Pearson GJ, Thanassoulis G, Anderson TJ, et al. 2021 Canadian Cardiovascular Society Guidelines for the Management of Dyslipidemia for the Prevention of Cardiovascular Disease in Adults. Can J Cardiol. 2021;37(8):1129-1150. doi:10.1016/j.cjca.2021.03.016
- Mora S, Otvos JD, Rifai N, Rosenson RS, Buring JE, Ridker PM. Lipoprotein particle profiles by nuclear magnetic resonance compared with standard lipids and apolipoproteins in predicting incident cardiovascular disease in women. Circulation. 2009;119(7):931-939. doi:10.1161/CIRCULATIONAHA.108.816181
- Williams KJ, Tabas I. The response-to-retention hypothesis of early atherogenesis. Arterioscler Thromb Vasc Biol. 1995;15(5):551-561. doi:10.1161/01.atv.15.5.551
- Tabas I, Williams KJ, Borén J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 2007;116(16):1832-1844. doi:10.1161/CIRCULATIONAHA.106.676890
- Borén J, Williams KJ. The central role of arterial retention of cholesterol-rich apolipoprotein-B-containing lipoproteins in the pathogenesis of atherosclerosis: a triumph of simplicity. Curr Opin Lipidol. 2016;27(5):473-483. doi:10.1097/MOL.0000000000000330
- Richardson TG, Sanderson E, Palmer TM, et al. Evaluating the relationship between circulating lipoprotein lipids and apolipoproteins with risk of coronary heart disease: A multivariable Mendelian randomisation analysis. PLoS Med. 2020;17(3):e1003062. Published 2020 Mar 23. doi:10.1371/journal.pmed.1003062
- Marston NA, Giugliano RP, Melloni GEM, et al. Association of Apolipoprotein B-Containing Lipoproteins and Risk of Myocardial Infarction in Individuals With and Without Atherosclerosis: Distinguishing Between Particle Concentration, Type, and Content. JAMA Cardiol. 2022;7(3):250-256. doi:10.1001/jamacardio.2021.5083
- Ference BA, Holmes MV, Smith GD. Using Mendelian Randomization to Improve the Design of Randomized Trials. Cold Spring Harb Perspect Med. 2021;11(7):a040980. Published 2021 Jul 1. doi:10.1101/cshperspect.a040980
- Sniderman AD, Williams K, Contois JH, et al. A meta-analysis of low-density lipoprotein cholesterol, non-high-density lipoprotein cholesterol, and apolipoprotein B as markers of cardiovascular risk. Circ Cardiovasc Qual Outcomes. 2011;4(3):337-345. doi:10.1161/CIRCOUTCOMES.110.959247
- Hindy G, Engström G, Larsson SC, et al. Role of Blood Lipids in the Development of Ischemic Stroke and its Subtypes: A Mendelian Randomization Study. Stroke. 2018;49(4):820-827. doi:10.1161/STROKEAHA.117.019653
- Allara E, Morani G, Carter P, et al. Genetic Determinants of Lipids and Cardiovascular Disease Outcomes: A Wide-Angled Mendelian Randomization Investigation. Circ Genom Precis Med. 2019;12(12):e002711. doi:10.1161/CIRCGEN.119.002711
- Amarenco P, Bogousslavsky J, Callahan A 3rd, et al. High-dose atorvastatin after stroke or transient ischemic attack. N Engl J Med. 2006;355(6):549-559. doi:10.1056/NEJMoa061894
- Sabatine MS, Giugliano RP, Keech AC, et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N Engl J Med. 2017;376(18):1713-1722. doi:10.1056/NEJMoa1615664
- Schwartz GG, Steg PG, Szarek M, et al. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N Engl J Med. 2018;379(22):2097-2107. doi:10.1056/NEJMoa1801174
- Sun L, Clarke R, Bennett D, et al. Causal associations of blood lipids with risk of ischemic stroke and intracerebral hemorrhage in Chinese adults. Nat Med. 2019;25(4):569-574. doi:10.1038/s41591-019-0366-x
- Bohula EA, Marston NA, Ruzza A, et al. Rationale and design of the effect of evolocumab in patients at high cardiovascular risk without prior myocardial infarction or stroke (VESALIUS-CV) trial. Am Heart J. 2024;269:179-190. doi:10.1016/j.ahj.2023.12.004
- Klarin D, Lynch J, Aragam K, et al. Genome-wide association study of peripheral artery disease in the Million Veteran Program. Nat Med. 2019;25(8):1274-1279. doi:10.1038/s41591-019-0492-5
- Bonaca MP, Nault P, Giugliano RP, et al. Low-Density Lipoprotein Cholesterol Lowering With Evolocumab and Outcomes in Patients With Peripheral Artery Disease: Insights From the FOURIER Trial (Further Cardiovascular Outcomes Research With PCSK9 Inhibition in Subjects With Elevated Risk). Circulation. 2018;137(4):338-350. doi:10.1161/CIRCULATIONAHA.117.032235
- Nissen SE, Lincoff AM, Brennan D, et al. Bempedoic Acid and Cardiovascular Outcomes in Statin-Intolerant Patients. N Engl J Med. 2023;388(15):1353-1364. doi:10.1056/NEJMoa2215024
- Harrison SC, Holmes MV, Burgess S, et al. Genetic Association of Lipids and Lipid Drug Targets With Abdominal Aortic Aneurysm: A Meta-analysis. JAMA Cardiol. 2018;3(1):26-33. doi:10.1001/jamacardio.2017.4293
- Thanassoulis G, Campbell CY, Owens DS, et al. Genetic associations with valvular calcification and aortic stenosis. N Engl J Med. 2013;368(6):503-512. doi:10.1056/NEJMoa1109034
- Kamstrup PR, Tybjærg-Hansen A, Nordestgaard BG. Elevated lipoprotein(a) and risk of aortic valve stenosis in the general population. J Am Coll Cardiol. 2014;63(5):470-477. doi:10.1016/j.jacc.2013.09.038
- Tsimikas S. A Test in Context: Lipoprotein(a): Diagnosis, Prognosis, Controversies, and Emerging Therapies. J Am Coll Cardiol. 2017;69(6):692-711. doi:10.1016/j.jacc.2016.11.042
- Tsimikas S, Karwatowska-Prokopczuk E, Gouni-Berthold I, et al. Lipoprotein(a) Reduction in Persons with Cardiovascular Disease. N Engl J Med. 2020;382(3):244-255. doi:10.1056/NEJMoa1905239
- Malick WA, Goonewardena SN, Koenig W, Rosenson RS. Clinical Trial Design for Lipoprotein(a)-Lowering Therapies: JACC Focus Seminar 2/3. J Am Coll Cardiol. 2023;81(16):1633-1645. doi:10.1016/j.jacc.2023.02.033
- O’Donoghue ML, Rosenson RS, Gencer B, et al. Small Interfering RNA to Reduce Lipoprotein(a) in Cardiovascular Disease. N Engl J Med. 2022;387(20):1855-1864. doi:10.1056/NEJMoa2211023
- Passey S, Jha J, Chanda A, Pillai A, Frishman WH, Aronow WS. Advances in RNA-Based Therapies Targeted at Lipoprotein(a): Olpasiran in the Management of Atherosclerotic Cardiovascular Disease. Cardiol Rev. Published online August 7, 2025. doi:10.1097/CRD.0000000000001017
- Doherty S, Hernandez S, Rikhi R, et al. Lipoprotein(a) as a Causal Risk Factor for Cardiovascular Disease. Curr Cardiovasc Risk Rep. 2025;19(1):8. doi:10.1007/s12170-025-00760-1
- Nicholls SJ, Nissen SE, Fleming C, et al. Muvalaplin, an Oral Small Molecule Inhibitor of Lipoprotein(a) Formation: A Randomized Clinical Trial. JAMA. 2023;330(11):1042-1053. doi:10.1001/jama.2023.16503
- Nordestgaard BG. Triglyceride-Rich Lipoproteins and Atherosclerotic Cardiovascular Disease: New Insights From Epidemiology, Genetics, and Biology. Circ Res. 2016;118(4):547-563. doi:10.1161/CIRCRESAHA.115.306249
- Pearson GJ, Thanassoulis G, Anderson TJ, et al. 2021 Canadian Cardiovascular Society Guidelines for the Management of Dyslipidemia for the Prevention of Cardiovascular Disease in Adults. Can J Cardiol. 2021;37(8):1129-1150. doi:10.1016/j.cjca.2021.03.016
- Blumenthal RS, Morris PB, Gaudino M, et al. 2026 ACC/AHA/AACVPR/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Dyslipidemia: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J Am Coll Cardiol. Published online March 13, 2026. doi:10.1016/j.jacc.2025.11.016
- Richardson TG, Wang Q, Sanderson E, et al. Effects of apolipoprotein B on lifespan and risks of major diseases including type 2 diabetes: a mendelian randomisation analysis using outcomes in first-degree relatives. Lancet Healthy Longev. 2021;2(6):e317-e326. doi:10.1016/S2666-7568(21)00086-6
- Brunham LR, Kruit JK, Pape TD, et al. Beta-cell ABCA1 influences insulin secretion, glucose homeostasis and response to thiazolidinedione treatment. Nat Med. 2007;13(3):340-347. doi:10.1038/nm1546
- Bhatt DL, Steg PG, Miller M, et al. Cardiovascular Risk Reduction with Icosapent Ethyl for Hypertriglyceridemia. N Engl J Med. 2019;380(1):11-22. doi:10.1056/NEJMoa1812792
- Colhoun HM, Betteridge DJ, Durrington PN, et al. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): multicentre randomised placebo-controlled trial. Lancet. 2004;364(9435):685-696. doi:10.1016/S0140-6736(04)16895-5
- Cromwell WC, Otvos JD, Keyes MJ, et al. LDL Particle Number and Risk of Future Cardiovascular Disease in the Framingham Offspring Study – Implications for LDL Management. J Clin Lipidol. 2007;1(6):583-592. doi:10.1016/j.jacl.2007.10.001
- Harada PHN, Demler OV, Dugani SB, et al. Lipoprotein insulin resistance score and risk of incident diabetes during extended follow-up of 20 years: The Women’s Health Study. J Clin Lipidol. 2017;11(5):1257-1267.e2. doi:10.1016/j.jacl.2017.06.008
- Lincoff AM, Brown-Frandsen K, Colhoun HM, et al. Semaglutide and Cardiovascular Outcomes in Obesity without Diabetes. N Engl J Med. 2023;389(24):2221-2232. doi:10.1056/NEJMoa2307563
- Bril F, Sninsky JJ, Baca AM, et al. Hepatic Steatosis and Insulin Resistance, But Not Steatohepatitis, Promote Atherogenic Dyslipidemia in NAFLD. J Clin Endocrinol Metab. 2016;101(2):644-652. doi:10.1210/jc.2015-3111
- Stefan N, Häring HU, Cusi K. Non-alcoholic fatty liver disease: causes, diagnosis, cardiometabolic consequences, and treatment strategies. Lancet Diabetes Endocrinol. 2019;7(4):313-324. doi:10.1016/S2213-8587(18)30154-2
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J Hepatol. 2016;64(6):1388-1402. doi:10.1016/j.jhep.2015.11.004
- Harrison SA, Bedossa P, Guy CD, et al. A Phase 3, Randomized, Controlled Trial of Resmetirom in NASH with Liver Fibrosis. N Engl J Med. 2024;390(6):497-509. doi:10.1056/NEJMoa2309000
- Romeo S, Kozlitina J, Xing C, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40(12):1461-1465. doi:10.1038/ng.257
- Kozlitina J, Smagris E, Stender S, et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2014;46(4):352-356. doi:10.1038/ng.2901
- Mikolasevic I, Žutelija M, Mavrinac V, Orlic L. Dyslipidemia in patients with chronic kidney disease: etiology and management. Int J Nephrol Renovasc Dis. 2017;10:35-45. Published 2017 Feb 7. doi:10.2147/IJNRD.S101808
- Baigent C, Landray MJ, Reith C, et al. The effects of lowering LDL cholesterol with simvastatin plus ezetimibe in patients with chronic kidney disease (Study of Heart and Renal Protection): a randomised placebo-controlled trial. Lancet. 2011;377(9784):2181-2192. doi:10.1016/S0140-6736(11)60739-3
- Fellström BC, Jardine AG, Schmieder RE, et al. Rosuvastatin and cardiovascular events in patients undergoing hemodialysis. N Engl J Med. 2009;360(14):1395-1407. doi:10.1056/NEJMoa0810177
- Lanktree MB, Thériault S, Walsh M, Paré G. HDL Cholesterol, LDL Cholesterol, and Triglycerides as Risk Factors for CKD: A Mendelian Randomization Study. Am J Kidney Dis. 2018;71(2):166-172. doi:10.1053/j.ajkd.2017.06.011
- Xie R, Sha S, Peng L, Holleczek B, Brenner H, Schöttker B. Metabolomics data improve 10-year cardiovascular risk prediction with the SCORE2 algorithm for the general population without cardiovascular disease or diabetes. Eur J Prev Cardiol. Published online April 24, 2025. doi:10.1093/eurjpc/zwaf254
- Lincoff AM, Brown-Frandsen K, Colhoun HM, et al. Semaglutide and Cardiovascular Outcomes in Obesity without Diabetes. N Engl J Med. 2023;389(24):2221-2232. doi:10.1056/NEJMoa2307563
- Welsh C, Celis-Morales CA, Brown R, et al. Comparison of Conventional Lipoprotein Tests and Apolipoproteins in the Prediction of Cardiovascular Disease. Circulation. 2019;140(7):542-552. doi:10.1161/CIRCULATIONAHA.119.041149
- Raal FJ, Rosenson RS, Reeskamp LF, et al. Evinacumab for Homozygous Familial Hypercholesterolemia. N Engl J Med. 2020;383(8):711-720. doi:10.1056/NEJMoa2004215
- Cuchel M, Raal FJ, Hegele RA, et al. 2023 Update on European Atherosclerosis Society Consensus Statement on Homozygous Familial Hypercholesterolaemia: new treatments and clinical guidance. Eur Heart J. 2023;44(25):2277-2291. doi:10.1093/eurheartj/ehad197
- Varbo A, Benn M, Tybjærg-Hansen A, Jørgensen AB, Frikke-Schmidt R, Nordestgaard BG. Remnant cholesterol as a causal risk factor for ischemic heart disease. J Am Coll Cardiol. 2013;61(4):427-436. doi:10.1016/j.jacc.2012.08.1026
- Ginsberg HN, Packard CJ, Chapman MJ, et al. Triglyceride-rich lipoproteins and their remnants: metabolic insights, role in atherosclerotic cardiovascular disease, and emerging therapeutic strategies-a consensus statement from the European Atherosclerosis Society. Eur Heart J. 2021;42(47):4791-4806. doi:10.1093/eurheartj/ehab551
- Das Pradhan A, Glynn RJ, Fruchart JC, et al. Triglyceride Lowering with Pemafibrate to Reduce Cardiovascular Risk. N Engl J Med. 2022;387(21):1923-1934. doi:10.1056/NEJMoa2210645
- Marston NA, Bergmark BA, Alexander VJ, et al. Olezarsen for Managing Severe Hypertriglyceridemia and Pancreatitis Risk. N Engl J Med. 2026;394(5):429-441. doi:10.1056/NEJMoa2512761
- Syed YY. Olezarsen: First Approval. Drugs. 2025;85(4):571-576. doi:10.1007/s40265-025-02166-0
- Wong TY, Cheung CM, Larsen M, Sharma S, Simó R. Diabetic retinopathy. Nat Rev Dis Primers. 2016;2:16012. Published 2016 Mar 17. doi:10.1038/nrdp.2016.12
- Keech AC, Mitchell P, Summanen PA, et al. Effect of fenofibrate on the need for laser treatment for diabetic retinopathy (FIELD study): a randomised controlled trial. Lancet. 2007;370(9600):1687-1697. doi:10.1016/S0140-6736(07)61607-9
- ACCORD Study Group; ACCORD Eye Study Group, Chew EY, et al. Effects of medical therapies on retinopathy progression in type 2 diabetes. N Engl J Med. 2010;363(3):233-244. doi:10.1056/NEJMoa1001288
- Preiss D, Logue J, Sammons E, et al. Effect of Fenofibrate on Progression of Diabetic Retinopathy. NEJM Evid. 2024;3(8):EVIDoa2400179. doi:10.1056/EVIDoa2400179
- Lim LS, Wong TY. Lipids and diabetic retinopathy. Expert Opin Biol Ther. 2012;12(1):93-105. doi:10.1517/14712598.2012.641531
- Mendola P, Ghassabian A, Mills JL, et al. Retinol-Binding Protein 4 and Lipids Prospectively Measured During Early to Mid-Pregnancy in Relation to Preeclampsia and Preterm Birth Risk. Am J Hypertens. 2017;30(6):569-576. doi:10.1093/ajh/hpx020
- Costantine MM, Cleary K, Hebert MF, et al. Safety and pharmacokinetics of pravastatin used for the prevention of preeclampsia in high-risk pregnant women: a pilot randomized controlled trial. Am J Obstet Gynecol. 2016;214(6):720.e1-720.e17. doi:10.1016/j.ajog.2015.12.038
- Akbar MIA, Azis MA, Riu DS, et al. INOVASIA Study: A Multicenter Randomized Clinical Trial of Pravastatin to Prevent Preeclampsia in High-Risk Patients. Am J Perinatol. 2024;41(9):1203-1211. doi:10.1055/a-1798-1925
- Pantoni L. Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. 2010;9(7):689-701. doi:10.1016/S1474-4422(10)70104-6
- Dichgans M, Leys D. Vascular Cognitive Impairment. Circ Res. 2017;120(3):573-591. doi:10.1161/CIRCRESAHA.116.308426
- Huang SY, Yang YX, Zhang YR, et al. Investigating Causal Relations Between Circulating Metabolites and Alzheimer’s Disease: A Mendelian Randomization Study. J Alzheimers Dis. 2022;87(1):463-477. doi:10.3233/JAD-220050
- Iwagami M, Qizilbash N, Gregson J, et al. Blood cholesterol and risk of dementia in more than 1·8 million people over two decades: a retrospective cohort study. Lancet Healthy Longev. 2021;2(8):e498-e506. doi:10.1016/S2666-7568(21)00150-1
- Westphal Filho FL, Moss Lopes PR, Menegaz de Almeida A, et al. Statin use and dementia risk: A systematic review and updated meta-analysis. Alzheimers Dement (N Y). 2025;11(1):e70039. Published 2025 Jan 16. doi:10.1002/trc2.70039
- Emanuele E, Peros E, Tomaino C, et al. Relation of apolipoprotein(a) size to alzheimer’s disease and vascular dementia. Dement Geriatr Cogn Disord. 2004;18(2):189-196. doi:10.1159/000079200
- Shamloul R, Ghanem H. Erectile dysfunction. Lancet. 2013;381(9861):153-165. doi:10.1016/S0140-6736(12)60520-0
- Cai X, Tian Y, Wu T, Cao CX, Bu SY, Wang KJ. The role of statins in erectile dysfunction: a systematic review and meta-analysis. Asian J Androl. 2014;16(3):461-466. doi:10.4103/1008-682X.123678
- Sofi F, Marcucci R, Fedi S, et al. High lipoprotein (a) levels are associated with an increased risk of retinal vein occlusion. Atherosclerosis. 2010;210(1):278-281. doi:10.1016/j.atherosclerosis.2009.11.006
- Kamstrup PR, Tybjærg-Hansen A, Nordestgaard BG. Genetic evidence that lipoprotein(a) associates with atherosclerotic stenosis rather than venous thrombosis. Arterioscler Thromb Vasc Biol. 2012;32(7):1732-1741. doi:10.1161/ATVBAHA.112.248765
- Poredoš P, Mukherjee D, Blinc A. Statins and Venous Thromboembolic Disease – Where are we Now?. Curr Vasc Pharmacol. 2024;22(4):297-300. doi:10.2174/0115701611308323240229050237
- Glynn RJ, Danielson E, Fonseca FA, et al. A randomized trial of rosuvastatin in the prevention of venous thromboembolism. N Engl J Med. 2009;360(18):1851-1861. doi:10.1056/NEJMoa0900241
- Yarmolinsky J, Wade KH, Richmond RC, et al. Causal Inference in Cancer Epidemiology: What Is the Role of Mendelian Randomization?. Cancer Epidemiol Biomarkers Prev. 2018;27(9):995-1010. doi:10.1158/1055-9965.EPI-17-1177
- Ray KK, Wright RS, Kallend D, et al. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N Engl J Med. 2020;382(16):1507-1519. doi:10.1056/NEJMoa1912387
- Mach F, Koskinas KC, Roeters van Lennep JE, et al. 2025 Focused Update of the 2019 ESC/EAS Guidelines for the management of dyslipidaemias. Eur Heart J. 2025;46(42):4359-4378. doi:10.1093/eurheartj/ehaf190
- Soffer DE, Marston NA, Maki KC, et al. Role of apolipoprotein B in the clinical management of cardiovascular risk in adults: An Expert Clinical Consensus from the National Lipid Association. J Clin Lipidol. 2024;18(5):e647-e663. doi:10.1016/j.jacl.2024.08.013
- Nidorf SM, Fiolet ATL, Mosterd A, et al. Colchicine in Patients with Chronic Coronary Disease. N Engl J Med. 2020;383(19):1838-1847. doi:10.1056/NEJMoa2021372
- Williams DM, Finan C, Schmidt AF, Burgess S, Hingorani AD. Lipid lowering and Alzheimer disease risk: A mendelian randomization study. Ann Neurol. 2020;87(1):30-39. doi:10.1002/ana.25642
Author note. This manuscript is intended for educational use on curingheartdisease.com and is not a substitute for individualized clinical advice. The author is a PhD researcher and not a licensed clinician. Citations are formatted in IEEE numerical style and intended to be verifiable in PubMed/CrossRef; readers are encouraged to consult the primary literature directly.