The Metabolic Economy of Human Cholesterol
Analysis of De Novo Synthesis, Lipoprotein Flux, and Peripheral Tissue Acquisition
1. Abstract
The physiological regulation of cholesterol in the human body is among the most sophisticated examples of metabolic homeostasis in mammalian biology. Cholesterol, a tetracyclic sterol, is indispensable for eukaryotic life-serving as an architectural keystone of cellular membranes, a substrate for steroid hormone and bile acid synthesis, and a scaffold for transmembrane signaling platforms. Peripheral tissues cannot cleave the sterol nucleus of cholesterol; hepatic conversion to bile acids is therefore the only bile-acid disposal route, while neutral sterol elimination also includes biliary and transintestinal excretion pathways.
Every nucleated cell possesses the enzymatic machinery to synthesize cholesterol de novo from acetyl-CoA via an approximately 30-step pathway. The high energetic cost of this synthesis drives preferential receptor-mediated LDL uptake in most tissues when circulating cholesterol is available. This report examines the quantitative distribution of cholesterol across human tissues, characterizes tissue-specific biosynthetic pathway utilization in light of definitive flux analysis data, and contextualizes the regulatory architecture that maintains sterol homeostasis.
2. Introduction
Cholesterol occupies a singular position in mammalian biochemistry. Unlike most metabolites that are either oxidized for energy or polymerized into macromolecular assemblies, cholesterol is managed as a durable structural asset. The sterol ring system is extraordinarily stable; no enzymatic pathway in human cells can cleave it to recover the carbon atoms as oxidizable fuel. Disposal is therefore an indirect, hepatic process involving conversion to bile acids (via CYP7A1 and subsidiary pathways) or direct excretion into bile [11].
The clinical importance of cholesterol is well-established. Elevated concentrations of LDL-cholesterol (LDL-C), and more precisely elevated LDL particle number as quantified by plasma apolipoprotein B (ApoB), are causally associated with atherosclerotic cardiovascular disease (ASCVD) across a broad base of Mendelian randomization, epidemiological, and interventional evidence [20]. A precise understanding of the underlying cholesterol economy has direct therapeutic implications: it explains why hyper-synthesizers respond robustly to HMG-CoA reductase inhibitors (statins) but poorly to dietary restriction, while hyper-absorbers show the converse pattern and benefit from intestinal NPC1L1 blockade with ezetimibe [28, 29]. It also explains why PCSK9 inhibition-by preventing post-translational LDL receptor (LDLR) degradation-amplifies hepatic LDL clearance independently of the mevalonate pathway [21].
3. The Quantitative Framework of Total Body Cholesterol
In a healthy adult human, total body cholesterol is approximately 2,100 mg per kilogram of body weight [6]. For a 70-kg individual, this corresponds to a pool of approximately 147 grams. Upwards of 90% of this pool resides within cell membranes, where cholesterol modulates bilayer fluidity and organizes cholesterol-enriched microdomains (‘lipid rafts’) that serve as platforms for receptor tyrosine kinases and G-protein-coupled receptors [19].
The daily net cholesterol balance reflects endogenous synthesis, dietary intake, the reabsorption of biliary cholesterol, and fecal loss of neutral and acidic sterols; it should not be reduced to a simple synthesis-versus-dietary absorption dichotomy. Human whole-body cholesterol turnover is approximately 0.7% per day-equivalent to roughly 0.8–1.2 grams per day in a 70-kg adult [5, 6]. In contrast, mice turn over approximately 7–9% of their total body cholesterol daily [5]-a difference with important implications for the translatability of rodent lipid-lowering studies to human outcomes.
Table 1. Daily cholesterol balance in a healthy adult under typical Western dietary conditions.
Note: This table presents established fluxes in the human cholesterol economy. Dietary absorption is not simply the ‘other side’ of de novo synthesis; it reflects a multistep intestinal process involving biliary secretion, luminal mixing, NPC1L1-mediated uptake, and partial biliary recirculation.
| Parameter | Typical Value / Range | Interpretive Note | Key Reference |
| Total body cholesterol | ~2,100 mg/kg body weight; ~147 g in 70-kg adult | Vast majority (>90%) in membranes; remainder in circulating lipoproteins | Goodman et al. 1973 [6] |
| Whole-body daily turnover | ~0.7%/day (~0.8–1.2 g/day in 70-kg adult) | Substantially lower than rodents (~7–9%/day); implies very conservative human sterol economy | Dietschy & Turley 2002 [5]; Goodman et al. 1973 [6] |
| De novo synthesis | 700–900 mg/day in healthy adults (substantial interindividual variability) | Not a simple counterweight to dietary absorption; reflects whole-body synthesis across all tissues | Grundy 1983 [3] |
| – Hepatic contribution | ~10–25% of total synthesis | Majority arises from extrahepatic tissues; exact fraction varies by phenotype and method | Dietschy et al. 1993 [4]; Dietschy & Turley 2002 [5] |
| Dietary cholesterol intake | Commonly ~200–500 mg/day (population- and epoch-dependent) | Population mean varies widely; not a primary driver of plasma LDL-C in most individuals due to compensatory SREBP2 suppression | Wang 2007 [9] |
| Intestinal cholesterol presented to lumen | ~1,400–1,700 mg/day (diet + bile + shed epithelial cells) | Biliary contribution dominates; dietary fraction is minority of luminal load | Wang 2007 [9]; Russell 2003 [11] |
| Fractional intestinal absorption | Highly variable: 29.0–81.1%, mean 56.2% in healthy subjects | Inter-individual variation is large; NPC1L1 genotype is a major determinant | Bosner et al. 1999 [10] |
| Biliary cholesterol secretion | ~800–1,200 mg/day | Primary luminal cholesterol source; most is reabsorbed; the fraction not reabsorbed is excreted as neutral sterol | Russell 2003 [11] |
| Bile acid synthesis | ~400 mg/day (approximate classic estimate) | Only bile-acid disposal route; biliary AND transintestinal pathways contribute to overall neutral sterol elimination | Russell 2003 [11] |
| Total fecal sterol loss | ~0.7–1.0 g/day (wide range) | Neutral sterols and acidic sterols should be distinguished; transintestinal cholesterol excretion (TICE) contributes in addition to biliary loss | Russell 2003 [11] |
| Steroid hormone synthesis | ~50 mg/day (rough historical estimate) | Weakly sourced; reported as order-of-magnitude approximation only | Miller & Auchus 2011 [12] |
All values represent physiological ranges under normal metabolic conditions. Values shift substantially under dyslipidaemia, liver disease, or pharmacological intervention. Exact hepatic synthesis share is method-dependent; the 10–25% range reflects human tracer data.
4. The Energetic Imperative: De Novo Synthesis as a Costly Asset
The complete pathway from acetyl-CoA to cholesterol comprises approximately 30 enzymatic steps [1]. It is divided into two major phases:
- The pre-lanosterol phase (~11 steps): encompasses the mevalonate pathway proper (6 dedicated enzymatic steps from acetyl-CoA to isopentenyl pyrophosphate [IPP]) through squalene synthesis and oxidative cyclization to lanosterol [1].
- The post-lanosterol phase (~19 steps): the conversion of lanosterol to cholesterol via divergent biosynthetic branches characterized by Bloch (1965) [1] and Kandutsch & Russell (1960) [17].
4.1 Bioenergetic Cost: A Nuanced Accounting
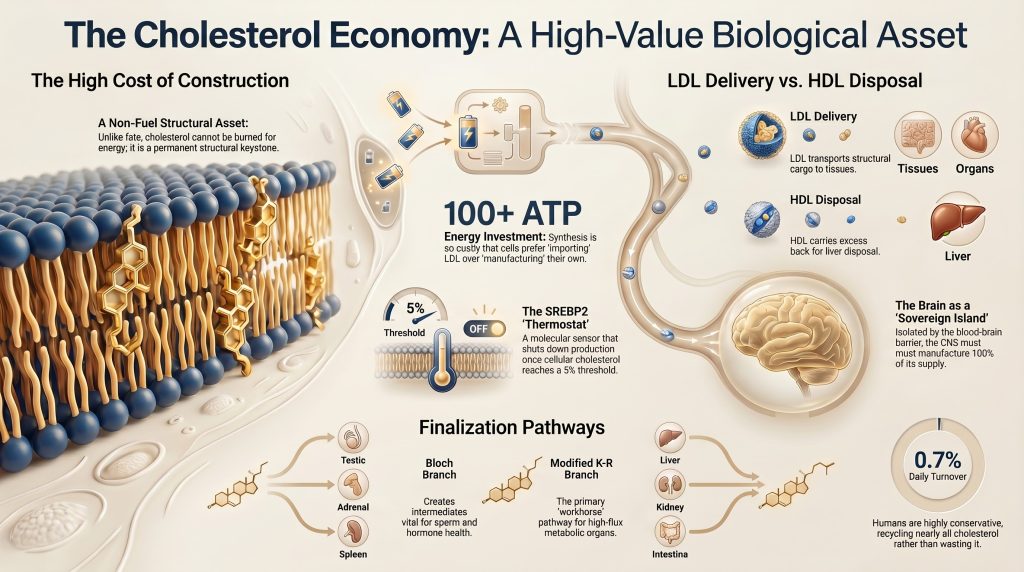
Cholesterol biosynthesis is energetically expensive. Commonly cited estimates are approximately 18 ATP and 16 NADPH per molecule from acetyl-CoA, accounting only for core pathway cofactors (mevalonate kinase steps and HMG-CoA reductase). These are the values recognized in most biochemistry textbook literature [1]. Broader accounting conventions-which additionally include the post-lanosterol NADPH consumed during the three oxidative demethylation steps and ring reductions of lanosterol to cholesterol-yield higher values (approximately 26 NADPH total across the full pathway [2]) there is no universally accepted single answer [2]. When the opportunity cost of 18 acetyl-CoA molecules (foregone β-oxidation energy) and all reducing equivalents are converted to ATP-equivalent units, the total investment per cholesterol molecule substantially exceeds 100 ATP-equivalents. This explains why cells strongly prefer receptor-mediated LDL uptake over de novo synthesis when circulating cholesterol is available [13].
4.2 PCSK9 and the LDL Receptor Cycle
The LDLR mediates receptor-mediated endocytosis of ApoB-100-containing LDL particles. After delivering its lipid payload to lysosomes, the receptor is recycled to the hepatocyte surface for subsequent rounds of LDL binding. Proprotein convertase subtilisin/kexin type 9 (PCSK9) disrupts this recycling by binding the LDLR in the endosome and directing it toward lysosomal degradation rather than surface return [21].
Loss-of-function variants in PCSK9 are associated with lifelong lower LDL-C and substantial reductions in coronary risk, but the magnitude of protection varies by variant type and population [21]. Monoclonal antibody inhibitors of PCSK9 (evolocumab, alirocumab) reduce LDL-C by 50–65% on top of statin therapy and significantly reduce major adverse cardiovascular events in high-risk patients [22, 23]. This mechanism is additive to statin-mediated HMG-CoA reductase inhibition and does not depend on the mevalonate pathway.
5. Pathway Variations: Bloch and Kandutsch–Russell Branches
Post-lanosterol cholesterol biosynthesis is better described as a spectrum of tissue-specific fluxes through Bloch-dominant and modified Kandutsch–Russell branches, rather than as a strict binary choice between canonical Bloch and canonical K-R pathways.
The tissue distribution of these pathways has been definitively characterized by Mitsche, McDonald, Hobbs, and Cohen [16] using stable-isotope flux analysis across 15 murine tissues in vivo. Their key finding is that canonical K-R was not observed as the dominant intact pathway in any tissue; instead, they described a modified K-R model in which liver and kidney showed predominantly modified K-R flux, while testis and adrenal were Bloch-heavy. Any summary that presents liver/kidney/intestine as sites of canonical K-R predominance as a resolved mammalian rule misrepresents both the authors’ conclusions and some specific tissue assignments.
5.1 Bloch Pathway (via Desmosterol)
The Bloch pathway proceeds through desmosterol (lanosterol → … → desmosterol → cholesterol via DHCR24). It predominates in tissues where the desmosterol intermediate itself serves a biological function. Testis are the clearest example: desmosterol accumulates in mature sperm heads, where its distinct biophysical properties relative to cholesterol facilitate membrane remodelling required for fertilisation competence [16, 18].
Bloch-predominant tissues in the Mitsche et al. analysis: testis, adrenal glands, and spleen. Separately, cell-type-specific studies in the brain demonstrate that astrocytes and developing neurons preferentially use Bloch/desmosterol synthesis. However, adult whole-brain tissue in the same Mitsche et al. analysis mapped predominantly to modified K-R flux. These two findings should not be conflated: adult whole-brain and specific neural cell-type evidence should be stated separately.
5.2 Modified Kandutsch–Russell Pathway (via 7-DHC / Lathosterol)
The modified K-R pathway proceeds through 7-dehydrocholesterol (7-DHC) and lathosterol (lanosterol → … → 7-DHC → cholesterol via DHCR7). Adult mouse liver in the Mitsche et al. analysis showed predominantly modified K-R flux. This is clinically important: DHCR7 deficiency causes Smith–Lemli–Opitz syndrome, in which toxic 7-DHC accumulation affects all modified K-R-dominant tissues [18]. In skin, 7-DHC is the immediate precursor of vitamin D₃ upon UVB irradiation, giving the modified K-R branch a distinctive photobiological role [18].
DHCR24 and DHCR7 are the terminal reductases that define important branch chemistry, but tissue flux is more plastic and branched than a strict binary pathway diagram suggests [16].
Table 2. Post-lanosterol biosynthetic pathway distribution by tissue (corrected per Mitsche et al. 2015 [16]).
| Pathway Branch | Tissues Where This Branch Predominates (in Mitsche et al. 2015 [16]) | Biological Rationale and Caveats |
| Bloch (via desmosterol) | Testis, adrenal glands, spleen; astrocytes and developing neurons (cell-type studies) | Desmosterol (the Bloch penultimate intermediate) has distinct biophysical properties important in spermatogenesis. Astrocyte-specific and developing-neuron studies indicate preferential Bloch/desmosterol flux. Adult whole-brain tissue, however, mapped predominantly to modified K-R flux in the Mitsche et al. mouse analysis |
| Modified Kandutsch–Russell (via 7-DHC/lathosterol) | Liver, kidney, preputial gland, intestine; adult whole-brain tissue (in Mitsche et al.) | Dominant in high-flux metabolic organs. Mitsche et al. described this as a ‘modified K-R’ pathway-canonical K-R was not observed as the dominant intact pathway in any tissue. The liver uses predominantly modified K-R flux, not Bloch. DHCR7 deficiency (Smith–Lemli–Opitz syndrome) demonstrates the clinical importance of this branch. |
| Mixed / context-dependent | Skin, skeletal muscle, heart, lung | Ratio shifts with developmental stage, oxygenation, and hormonal context. In skin, K-R intermediate 7-DHC is the direct photochemical precursor of vitamin D₃. Tissue flux is better described as a plastic spectrum than a strict binary choice. |
Data derive from stable-isotope flux analysis in 15 murine tissues [16]. Human tissue distribution is inferred by enzyme homology; ‘modified K-R’ refers to Mitsche et al.’s own terminology-canonical K-R was not observed as the predominant intact pathway in any tissue.
6. Lipoprotein Dynamics: The Systemic Logistics System
Given that cells can synthesize their own cholesterol, circulating lipoproteins serve three primary functions: (1) resource redistribution from synthesis-surplus to synthesis-deficient tissues; (2) metabolic buffering to spare cellular ATP and NADPH during high-energy-demand states; and (3) reverse transport and disposal of excess peripheral sterols, preventing accumulation in macrophages and atherogenesis [25]. Cholesterol is highly lipophilic and insoluble in aqueous plasma; it must be packaged into amphipathic lipoprotein particles alongside phospholipids and apolipoproteins that confer receptor-binding specificity [30].
6.1 The VLDL–IDL–LDL Cascade
VLDL particles are assembled in hepatocytes and secreted into the hepatic venous (systemic) circulation, where lipoprotein lipase (LPL) anchored to endothelial surfaces progressively hydrolyzes the triglyceride core, remodelling VLDL through intermediate-density lipoprotein (IDL) remnants until the triglyceride-depleted, cholesteryl ester-enriched LDL particle remains [30]. Each LDL particle carries a single ApoB-100 molecule; therefore, plasma ApoB concentration is a more precise index of atherogenic particle burden than LDL-cholesterol mass, which conflates particle size heterogeneity [20]. Approximately 60–70% of circulating cholesterol mass is carried within the LDL fraction [30].
6.2 HDL and Reverse Cholesterol Transport
High-density lipoprotein particles facilitate the return of excess peripheral cholesterol to the liver via reverse cholesterol transport (RCT). ABCA1 mediates the initial transfer of cellular cholesterol and phospholipid to lipid-poor apoA-I, forming nascent discoidal HDL particles. ABCG1 promotes further cholesterol efflux mainly to more mature, lipid-rich HDL particles; these are distinct initial acceptor pools with non-overlapping functional roles [26]. Lecithin-cholesterol acyltransferase (LCAT), activated by ApoA-I, then esterifies acquired cholesterol, driving net efflux thermodynamically and converting nascent discs into mature spherical HDL [26].
Cholesterol is returned to the liver via SR-BI–mediated selective uptake or via CETP-mediated exchange into ApoB-containing particles subsequently cleared by LDLR. The liver converts returned cholesterol into bile acids (primary: cholic acid, chenodeoxycholic acid) via CYP7A1, or excretes it directly into bile for neutral sterol elimination [11]. Transintestinal cholesterol excretion (TICE) provides an additional fecal disposal route that does not depend on biliary secretion.
6.3 SR-BI and Selective Uptake in Steroidogenic Tissues
SR-BI (scavenger receptor class B type I) mediates selective uptake of cholesteryl esters from both HDL and LDL without concomitant degradation of the lipoprotein particle. SR-BI is highly expressed in adrenal cortex and gonads and is upregulated by ACTH in the adrenal, creating a feedforward mechanism coupling hormonal demand to circulating sterol supply [24]. However, SR-BI predominance as the steroidogenic uptake pathway is better established in rodent adrenal than in human adrenal; in humans, LDLR-mediated LDL uptake remains especially important and the HDL–SR-BI pathway appears less dominant than in rodents. Both pathways contribute, and overgeneralization from rodent SR-BI data to humans should be avoided.
7. Tissue-Specific Cholesterol Acquisition Profiles
The degree to which a tissue relies on circulating cholesterol versus internal synthesis reflects its metabolic role, vascular access, and biosynthetic capacity. Table 3 below provides qualitative tissue profiles; specific import-vs-synthesis percentages for most peripheral tissues (muscle, adipose, skin) are not established by direct human tracer evidence and should not be stated as settled figures.
Table 3. Qualitative tissue cholesterol acquisition profiles.
| Tissue | Cholesterol Acquisition Profile and Functional Role |
| Brain (CNS) | Essentially self-sufficient for cholesterol under normal adult conditions; plasma lipoprotein-cholesterol influx across the blood–brain barrier is negligible. All cholesterol is synthesized locally, primarily by astrocytes, and distributed to neurons via ApoE-containing CSF lipoproteins. Precise synthesis/uptake split cannot be reliably quantified by tissue-fraction percentages in humans. |
| Adrenal cortex | Major dependence on plasma lipoprotein-derived cholesterol; in humans, LDLR-mediated LDL uptake is especially important, with SR-BI also relevant (but SR-BI predominance is better established in rodent adrenal than human adrenal). Under acute ACTH stimulation, >80% of steroid precursor cholesterol is derived from circulating lipoproteins. Intracellular cholesterol ester stores (regulated by ACAT/HSL) provide a buffer. |
| Liver | Central hub: performs receptor-mediated LDL uptake, de novo synthesis, VLDL secretion, bile acid synthesis, and biliary excretion. Exact fractional contribution to whole-body de novo synthesis varies by source and method (~10–25% in human steady-state data). Uses predominantly modified K-R biosynthetic flux. Precise import-vs-synthesis percentages are method-dependent. |
| Intestine | Major organ for both cholesterol absorption (NPC1L1-mediated from lumen) and de novo synthesis. Also participates in cholesterol excretion via transintestinal cholesterol excretion (TICE), which contributes to fecal sterol loss independently of biliary secretion. Predominantly modified K-R biosynthetic flux. |
| Gonads | Use a mixture of de novo synthesis, intracellular cholesterol ester reserves (mobilized by HSL), and plasma lipoproteins (both LDLR and SR-BI pathways). Exact percentages are context-dependent and acutely regulated by gonadotropins (LH/FSH). Bloch pathway dominates in testis. |
| Skeletal muscle, adipose, skin | Important to whole-body cholesterol economy by sheer mass, but precise human import-vs-synthesis ratios are not established without direct tracer evidence. Broad estimates suggest predominant reliance on local synthesis under normal conditions, but these should not be assigned specific percentages without citing the primary tracer study that supports them in humans. |
Qualitative descriptions reflect the consensus from human and animal studies. Precise percentage assignments for peripheral tissues (skeletal muscle, adipose, skin) are not established by direct human tracer evidence; see Dietschy et al. 1993 [4] and Goodman et al. 1973 [6] for available primary kinetic data. Tissue flux varies substantially under inflammation, metabolic disease, hypoxia, and pharmacological intervention.
7.1 Brain: Self-Sufficiency
The CNS contains approximately 20–25% of total body cholesterol despite comprising only ~2% of body mass [7]. The blood–brain barrier (BBB) formed by tight junctions between cerebral endothelial cells excludes lipoprotein-bound cholesterol from the systemic circulation; net influx of plasma cholesterol across the adult BBB is negligible [7, 8]. The brain must therefore synthesize all its own cholesterol, a task performed primarily by astrocytes, which transfer cholesterol to neurons via ApoE-containing lipoproteins in the cerebrospinal fluid [8].
Brain cholesterol is eliminated mainly by conversion to oxysterols, especially 24(S)-hydroxycholesterol, via neuronal CYP46A1 (cholesterol 24-hydroxylase). Direct jugular-venous arteriovenous difference measurements suggest a brain export of approximately 2–3 mg/day; older indirect estimates suggested 4–7 mg/day. This is an estimate range with method dependence-not a fixed single value [8]. Myelin sheaths are the most cholesterol-rich membrane assemblies in mammalian biology; cholesterol constitutes approximately 25–40% of total myelin lipid (the exact fraction is species-, region-, and measurement-dependent) [7].
7.2 Steroidogenic Tissues: Import-Dependent High-Flux Consumers
The adrenal cortex and gonads require cholesterol as the exclusive substrate for steroid hormone synthesis via the StAR protein-mediated mitochondrial import pathway [12]. Under acute ACTH stimulation, more than 80% of the cholesterol consumed in steroidogenesis is derived from circulating lipoproteins [24]. Intracellular cholesterol ester stores (ACAT-mediated esterification, HSL-mediated mobilization) provide a rapidly accessible buffer.
7.3 Skeletal Muscle, Adipose Tissue, and Skin
These tissues collectively represent the largest mass in the body and therefore a large absolute fraction of the cholesterol pool, yet their daily fractional turnover is low and most cholesterol is synthesized locally. Broad estimates from animal tracer studies suggest high reliance on de novo synthesis under normal conditions, but human in-vivo import-vs-synthesis percentages for these tissues are not established with the precision that a table of specific numbers implies. Their contribution to systemic lipid metabolism is also indirect: higher skeletal muscle mass is epidemiologically associated with lower circulating LDL-C, potentially via elevated LPL activity, improved insulin sensitivity (which upregulates hepatic LDLR expression), or myokine-mediated effects on hepatic lipoprotein metabolism [33, 34].
8. The Functional Paradox: Why Cholesterol Is Not a Fuel
A critical conceptual distinction in lipid metabolism is that while triglycerides are the primary lipid energy reserve and are catabolized via β-oxidation for ATP generation, cholesterol is categorically non-fuel. The tetracyclic sterol ring system is extraordinarily stable and lacks the high-energy C–H bonds of fatty acyl chains. Human cells possess no enzymatic pathway for sterol ring cleavage; the carbon atoms of cholesterol cannot be recovered as acetyl-CoA units for the TCA cycle [1].
Circulating lipoproteins therefore serve three non-caloric functions:
- Structural maintenance: The primary destination for circulating cholesterol is the plasma membrane. Cholesterol intercalates between phospholipid fatty acyl chains, condensing the bilayer and organizing liquid-ordered (Lo) microdomains (‘lipid rafts’) that serve as platforms for signaling receptors [19]. This function is membrane-density-dependent and cannot be performed by triglycerides.
- Precursor distribution: Circulating LDL provides the immediate sterol substrate for adrenocortical steroidogenesis. By maintaining a systemic LDL pool, the body ensures cortisol and aldosterone production can be initiated within minutes of ACTH stimulation, without waiting for de novo synthesis [12, 24].
- Homeostatic buffering: During tissue injury or rapid proliferation, cells can recruit circulating cholesterol for emergency membrane biogenesis, conserving ATP and NADPH at a time of maximal metabolic demand [13].
8.1 Mitochondrial Cholesterol Accumulation: Pathological Consequences
While cholesterol is indispensable in the plasma membrane, its accumulation in the inner mitochondrial membrane (IMM) above physiological concentrations is pathological. The IMM is normally the most cholesterol-depleted membrane in the cell-a condition required to maintain the fluidity and cardiolipin content necessary for optimal respiratory chain function [27]. Excess IMM cholesterol impairs complex I and complex III activity, reduces the mitochondrial membrane potential, and diminishes mitochondrial glutathione import, increasing vulnerability to reactive oxygen species generated by partially coupled respiratory chain activity [27]. This mechanism has been implicated in the steatohepatitis-to-cirrhosis transition in non-alcoholic fatty liver disease and in the vulnerability of foam cells in atherosclerotic plaques [27].
Two caveats on precision: (a) the exact pathological threshold for IMM cholesterol accumulation is not established clearly enough for a decisive human numerical statement-qualitative description is more defensible than citing specific mol% thresholds as settled fact; and (b) the identity of the mitochondrial glutathione transporter involved is debated-a direct reconstitution study challenged OGC/DIC-mediated GSH transport-and this is to date uncertain.
9. Regulatory Feedback: The SREBP2 Hub
Cellular cholesterol sensing is centered on Sterol Regulatory Element-Binding Protein 2 (SREBP2), a membrane-bound transcription factor in the endoplasmic reticulum (ER) [14]. ER cholesterol is maintained at low levels relative to the plasma membrane under normal conditions; SREBP-2 processing falls abruptly when ER cholesterol approaches approximately 5 mol% of ER membrane lipid [15].
When ER cholesterol falls below this threshold, the SCAP–SREBP2 complex dissociates from INSIG-1/INSIG-2 retention proteins, allowing SCAP to escort SREBP2 to the Golgi, where site-1 and site-2 proteases sequentially cleave SREBP2, releasing the N-terminal transcription factor domain for nuclear import [14]. In the nucleus, SREBP2 drives coordinated upregulation of:
- HMGCR (HMG-CoA reductase, the rate-limiting enzyme of the mevalonate pathway): to increase internal synthesis.
- LDLR (LDL receptor): to increase receptor-mediated LDL uptake from blood.
- PCSK9: paradoxically upregulated by SREBP2 as a negative feedback limiter on LDLR recycling-the mechanistic basis for PCSK9 inhibitor pharmacology [21].
When ER cholesterol rises to approximately 5 mol%-a switch-like threshold established by Radhakrishnan et al. [15] using quantitative membrane biochemistry-SCAP is retained in the ER by INSIG, SREBP2 remains inactive, and HMG-CoA reductase is targeted for proteasomal degradation via INSIG-mediated recruitment of gp78 E3 ubiquitin ligase [14, 15]. This bistable switch ensures cells exploit the circulating pool only when necessary.
10. Inter-Individual Variability and Clinical Implications
10.1 Hyper-Synthesizers versus Hyper-Absorbers
Individuals differ substantially in the relative contribution of synthesis versus absorption to their circulating cholesterol pool-a clinically actionable distinction [28]. ‘Hyper-synthesizers’ carry elevated plasma lathosterol (a synthesis marker) and depressed campesterol/sitosterol (absorption markers) relative to population medians. Statins produce robust LDL-C reductions in these patients. Conversely, ‘hyper-absorbers’ display the opposite plasma sterol profile; their intestinal NPC1L1 transporter captures a disproportionate fraction of both dietary and biliary cholesterol. In hyper-absorbers, statins may produce attenuated LDL-C reductions because compensatory NPC1L1 upregulation partially offsets synthesis inhibition; ezetimibe combination therapy restores a more complete response [28, 29].
The hyper-synthesizer/hyper-absorber framework is biologically useful and supported by non-cholesterol sterol biomarker data, but it is not yet a universally standardized, guideline-mandated clinical taxonomy: higher absorption markers do not always predict differential LDL-C responses, especially in FH cohorts. The framework should be presented as helpful but not deterministic.
10.2 Familial Hypercholesterolaemia and the LDLR Pathway
Familial hypercholesterolemia (FH) is the most common serious monogenic disorder in humans; the prevalence is approximately 1 in 250–300 (a recent NLA consensus estimate places global prevalence near 1 in 311, depending on population and diagnostic definition) [21]. FH arises from loss-of-function mutations in LDLR, gain-of-function mutations in PCSK9, or pathogenic variants in APOB, all of which impair receptor-mediated LDL clearance [21]. Because hepatic LDL uptake is reduced, SREBP2 remains constitutively active, driving upregulation of HMGCR and paradoxically increasing hepatic synthesis in the context of impaired exogenous uptake [14]-a vicious cycle that, untreated, leads to premature ASCVD.
11. Oxysterols: The Regulatory Layer Within the Cholesterol Economy
Oxysterols are oxidized cholesterol derivatives present in tissues and plasma at concentrations 3–4 orders of magnitude lower than cholesterol itself, yet they exert disproportionate regulatory influence on sterol homeostasis and immune function [31, 32].
Several oxysterols function as endogenous INSIG ligands and as activators of liver X receptors (LXRs), nuclear receptors that transcriptionally upregulate ABCA1, ABCG1, and ApoE-the machinery of reverse cholesterol transport [31, 32]. LXR activation by oxysterols (including 24(S),25-epoxycholesterol and 27-hydroxycholesterol) thus creates a feedforward coupling between cellular sterol excess and its removal. In the brain, 24(S)-hydroxycholesterol produced by neuronal CYP46A1 not only serves the cholesterol export function described in Section 7.1 but also acts as a positive allosteric modulator of NMDA-type glutamate receptors, linking cholesterol turnover to synaptic plasticity [8].
12. Limitations
Several fundamental questions in human cholesterol metabolism remain incompletely resolved and one must be careful to interpret the data with this in mind.
- The exact hepatic fraction of total whole-body de novo cholesterol synthesis. Available human tracer data suggest ~10–25%, but this is phenotype-dependent and technically difficult to measure in vivo.
- The tissue-specific ratio of Bloch-to-modified-K-R flux in humans across the lifespan. The Mitsche et al. 2015 study was performed in mice; human direct data are limited.
- The exact identity of the mitochondrial glutathione transporter involved in cholesterol-related oxidative stress; current evidence is contested.
- The pathological IMM cholesterol threshold in humans; precise mol% thresholds established in model systems may not translate directly to the clinical setting.
- The mechanism linking skeletal muscle mass to circulating LDL-C-whether via LPL activity, myokine signaling, or insulin sensitivity-mediated LDLR upregulation [33, 34].
- Transintestinal cholesterol excretion (TICE) as a quantitatively significant fecal sterol disposal pathway; its contribution relative to biliary excretion under various physiological and pharmacological conditions requires further human characterization.
13. Conclusion
The human cholesterol economy is a dual system of local self-sufficiency and systemic interdependence. Most cells synthesize cholesterol de novo via an approximately 30-step pathway at a substantial metabolic cost (~18 ATP and ~16 NADPH by the core cofactor accounting; higher under broader accounting conventions). This cost drives preferential receptor-mediated LDL uptake in most peripheral tissues when circulating cholesterol is available.
Circulating lipoproteins do not carry cholesterol as a fuel. LDL acts as a mobile reservoir of structural and steroidogenic precursor material; HDL facilitates the obligatory return of peripheral sterols to the liver via reverse cholesterol transport-the only route by which the sterol nucleus can ultimately be eliminated from the body.
Post-lanosterol biosynthesis is more accurately described as a tissue-specific flux spectrum than a strict Bloch-versus-K-R binary. The liver uses predominantly modified Kandutsch–Russell flux; the testis and adrenal use predominantly Bloch flux; adult whole-brain maps predominantly to modified K-R in flux analysis, while astrocytes and developing neurons show stronger Bloch/desmosterol signatures that should not be conflated with the whole-organ picture. Precise numerical percentages for tissue import-vs-synthesis ratios in humans are not fully understood for most peripheral tissues.
A clinically actionable understanding of this economy-encompassing the SREBP2 switch, the hyper-synthesizer/hyper-absorber spectrum, the PCSK9/LDLR axis, and the oxysterol regulatory layer-underpins rational selection among statins, ezetimibe, PCSK9 inhibitors, and emerging agents targeting de novo synthesis and reverse transport.
References
- Bloch K. The biological synthesis of cholesterol. Science. 1965;150(3692):19-28. doi:10.1126/science.150.3692.19
- Gaylor JL. Membrane-bound enzymes of cholesterol synthesis from lanosterol. Biochem Biophys Res Commun. 2002;292(5):1139-1146. doi:10.1006/bbrc.2001.2008
- Grundy SM. Absorption and metabolism of dietary cholesterol. Annu Rev Nutr. 1983;3:71-96. doi:10.1146/annurev.nu.03.070183.000443
- Dietschy JM, Turley SD, Spady DK. Role of liver in the maintenance of cholesterol and low density lipoprotein homeostasis in different animal species, including humans. J Lipid Res. 1993;34(10):1637-1659.
- Dietschy JM, Turley SD. Control of cholesterol turnover in the mouse. J Biol Chem. 2002;277(6):3801-3804. doi:10.1074/jbc.R100057200
- Goodman DS, Noble RP, Dell RB. The effects of colestipol resin and of colestipol plus clofibrate on the turnover of plasma cholesterol in man. J Clin Invest. 1973;52(10):2646-2655. doi:10.1172/JCI107457
- Dietschy JM, Turley SD. Thematic review series: brain Lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J Lipid Res. 2004;45(8):1375-1397. doi:10.1194/jlr.R400004-JLR200
- Björkhem I, Meaney S. Brain cholesterol: long secret life behind a barrier. Arterioscler Thromb Vasc Biol. 2004;24(5):806-815. doi:10.1161/01.ATV.0000120374.59826.1b
- Wang DQ. Regulation of intestinal cholesterol absorption. Annu Rev Physiol. 2007;69:221-248. doi:10.1146/annurev.physiol.69.031905.160725
- Bosner MS, Lange LG, Stenson WF, Ostlund RE Jr. Percent cholesterol absorption in normal women and men quantified with dual stable isotopic tracers and negative ion mass spectrometry. J Lipid Res. 1999;40(2):302-308.
- Russell DW. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem. 2003;72:137-174. doi:10.1146/annurev.biochem.72.121801.161712
- Miller WL, Auchus RJ. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr Rev. 2011;32(1):81-151. doi:10.1210/er.2010-0013
- Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986;232(4746):34-47. doi:10.1126/science.3513311
- Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89(3):331-340. doi:10.1016/s0092-8674(00)80213-5
- Radhakrishnan A, Goldstein JL, McDonald JG, Brown MS. Switch-like control of SREBP-2 transport triggered by small changes in ER cholesterol: a delicate balance. Cell Metab. 2008;8(6):512-521. doi:10.1016/j.cmet.2008.10.008
- Mitsche MA, McDonald JG, Hobbs HH, Cohen JC. Flux analysis of cholesterol biosynthesis in vivo reveals multiple tissue and cell-type specific pathways. Elife. 2015;4:e07999. Published 2015 Jun 26. doi:10.7554/eLife.07999
- KANDUTSCH AA, RUSSELL AE. Preputial gland tumor sterols. 3. A metabolic pathway from lanosterol to cholesterol. J Biol Chem. 1960;235:2256-2261.
- Porter FD, Herman GE. Malformation syndromes caused by disorders of cholesterol synthesis. J Lipid Res. 2011;52(1):6-34. doi:10.1194/jlr.R009548
- Simons K, Vaz WL. Model systems, lipid rafts, and cell membranes. Annu Rev Biophys Biomol Struct. 2004;33:269-295. doi:10.1146/annurev.biophys.32.110601.141803
- Ference BA, Ginsberg HN, Graham I, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2017;38(32):2459-2472. doi:10.1093/eurheartj/ehx144
- Abifadel M, Varret M, Rabès JP, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34(2):154-156. doi:10.1038/ng1161
- Sabatine MS, Giugliano RP, Keech AC, et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N Engl J Med. 2017;376(18):1713-1722. doi:10.1056/NEJMoa1615664
- Schwartz GG, Steg PG, Szarek M, et al. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N Engl J Med. 2018;379(22):2097-2107. doi:10.1056/NEJMoa1801174
- Glass C, Pittman RC, Weinstein DB, Steinberg D. Dissociation of tissue uptake of cholesterol ester from that of apoprotein A-I of rat plasma high density lipoprotein: selective delivery of cholesterol ester to liver, adrenal, and gonad. Proc Natl Acad Sci U S A. 1983;80(17):5435-5439. doi:10.1073/pnas.80.17.5435
- Tall AR. Cholesterol efflux pathways and other potential mechanisms involved in the athero-protective effect of high density lipoproteins. J Intern Med. 2008;263(3):256-273. doi:10.1111/j.1365-2796.2007.01898.x
- Rye KA, Barter PJ. Formation and metabolism of prebeta-migrating, lipid-poor apolipoprotein A-I. Arterioscler Thromb Vasc Biol. 2004;24(3):421-428. doi:10.1161/01.ATV.0000104029.74961.f5
- Garcia-Ruiz C, Mari M, Colell A, et al. Mitochondrial cholesterol in health and disease. Histol Histopathol. 2009;24(1):117-132. doi:10.14670/HH-24.117
- Miettinen TA, Gylling H. Cholesterol absorption efficiency and sterol metabolism in obesity. Atherosclerosis. 2000;153(1):241-248. doi:10.1016/s0021-9150(00)00404-4
- Sudhop T, Lütjohann D, Kodal A, et al. Inhibition of intestinal cholesterol absorption by ezetimibe in humans. Circulation. 2002;106(15):1943-1948. doi:10.1161/01.cir.0000034044.95911.dc
- Goldstein JL, Brown MS. A century of cholesterol and coronaries: from plaques to genes to statins. Cell. 2015;161(1):161-172. doi:10.1016/j.cell.2015.01.036
- Janowski BA, Willy PJ, Devi TR, Falck JR, Mangelsdorf DJ. An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature. 1996;383(6602):728-731. doi:10.1038/383728a0
- Kalaany NY, Mangelsdorf DJ. LXRS and FXR: the yin and yang of cholesterol and fat metabolism. Annu Rev Physiol. 2006;68:159-191. doi:10.1146/annurev.physiol.68.033104.152158
- Vella CA, Nelson MC, Unkart JT, Miljkovic I, Allison MA. Skeletal muscle area and density are associated with lipid and lipoprotein cholesterol levels: The Multi-Ethnic Study of Atherosclerosis. J Clin Lipidol. 2020;14(1):143-153. doi:10.1016/j.jacl.2020.01.002
- Hong S, Chang Y, Jung HS, Yun KE, Shin H, Ryu S. Relative muscle mass and the risk of incident type 2 diabetes: A cohort study. PLoS One. 2017;12(11):e0188650. Published 2017 Nov 30. doi:10.1371/journal.pone.0188650