Cholesterol Structure, Transport, and Biological Requirement
A mechanistic analysis of compartment partitioning, lipoprotein structural requirements, and the model-estimated minimum for the circulating transport system
Abstract
Background. Whole-body cholesterol distribution, lipoprotein structural requirements, and tissue dependence on circulating cholesterol are frequently conflated in clinical and educational sources, leading to overstatements of how much circulating cholesterol the body “requires.”
Methods. This analysis (i) defines a strict compartment-based vocabulary distinguishing total body cholesterol, tissue cholesterol, intracellular cholesterol, and circulating (plasma/serum) cholesterol, and (ii) builds a transparent transport-model estimate for the minimum circulating cholesterol concentration required to maintain lipoprotein structural integrity and to meet exogenous tissue demand, with stated assumptions, explicit unit-converted parameters, and sensitivity bounds. Equations and parameter ranges are given in the Methods appendix (Section 11).
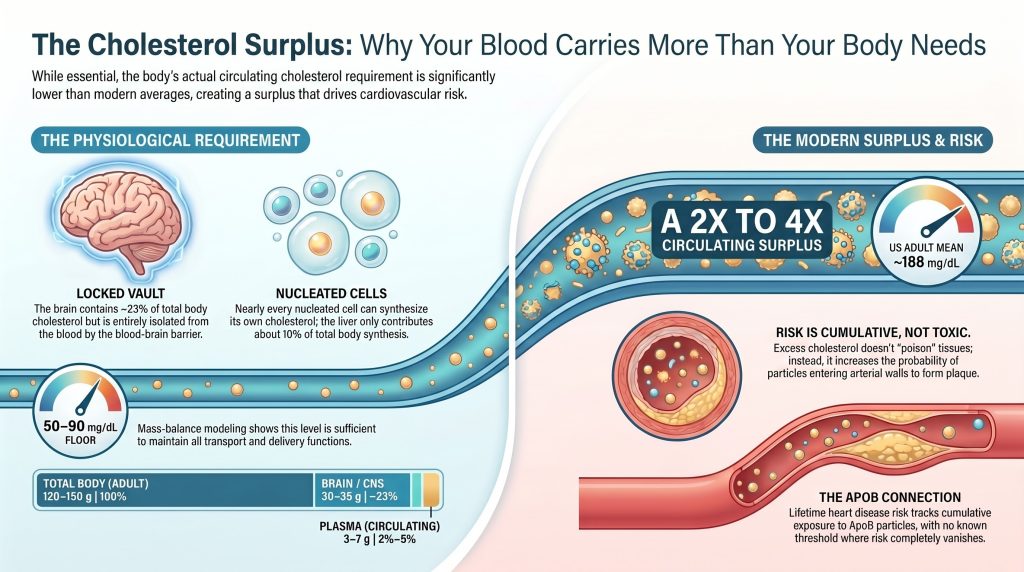
Findings. (1) Total body cholesterol in an adult is approximately 120–150 g; the rapidly-exchanging plasma pool of 3–7 g (depending on plasma volume and measured total cholesterol concentration) represents 2–5% of body content. (2) The brain alone contains ~30–35 g, isolated from plasma by the blood–brain barrier. (3) Plasma cholesterol is ~70% esterified / ~30% free. (4) A representative 22-nm modeled LDL particle (Hevonoja reconstruction) contains ~2,200 cholesterol molecules, ~18% of which is surface free cholesterol; LDL is heterogeneous and these numbers should not be applied uncritically to all LDL particles. (5) Under stated transport-model assumptions and using NMR-consistent particle counts (LDL-P in nmol/L → ~10¹⁶–10¹⁷ particles/dL; HDL-P in µmol/L → ~10¹⁸ particles/dL), the model-estimated minimum plasma total cholesterol that simultaneously stabilizes the lipoprotein system and supplies ~50 mg/day of net exogenous demand in a nonpregnant adult is approximately 50–90 mg/dL (sensitivity 40–110 mg/dL). This is a calculated scenario, not a demonstrated physiological minimum.
Conclusions. Modern US adult mean plasma total cholesterol of ~188 mg/dL (NHANES 2017–2018) exceeds the model-estimated transport floor by a factor that is assumption-sensitive but consistently > 2. The surplus is metabolically tolerated, causally tied to ASCVD risk on a per-apoB-particle basis, and not required for any known physiological function. Clinical guidelines that recommend LDL-C reduction for ASCVD prevention operate in a different epistemic register (event reduction in trials) than this paper (mass-balance modeling); the two are complementary, not in conflict.
1. Compartment-Based Vocabulary
Every cholesterol claim in this paper specifies (i) the compartment, (ii) the chemical form, and (iii) the units. Conflating clinical lipid-panel concentrations with tissue or whole-body cholesterol mass is the source of most quantitative errors in the lay and educational literature. The following terminology is used throughout. [1]
| Term | Meaning | Typical units |
| Plasma TC / serum TC | Total cholesterol concentration in the corresponding blood fraction (calculated or directly measured on a routine lipid panel) | mg/dL or mmol/L |
| Plasma LDL-C / HDL-C / VLDL-C | Cholesterol carried by the named lipoprotein class in plasma | mg/dL or mmol/L |
| LDL-P / HDL-P | Number concentration of LDL or HDL particles (typically measured by NMR spectroscopy) | LDL-P in nmol/L; HDL-P in µmol/L (note different orders of magnitude) |
| Plasma free cholesterol / plasma cholesteryl ester | Chemical partition of plasma cholesterol between unesterified and esterified forms (typically ~30% / ~70%) | mg/dL, mmol/L, or % of plasma TC |
| Plasma cholesterol mass | Plasma TC × plasma volume; the absolute cholesterol mass circulating in plasma at one moment | g (= mg/dL × dL_plasma / 1000) |
| Circulating cholesterol mass | Plasma cholesterol mass + cholesterol carried in erythrocyte membranes (rapidly exchanges with plasma) | g |
| Tissue cholesterol concentration | Cholesterol per unit mass of tissue (e.g., brain at ~23 mg/g wet weight) | mg/g wet or dry weight |
| Cellular / subcellular cholesterol | Cholesterol in a specific cell type, organelle membrane, or pool | mol% of membrane lipids; µg/mg protein |
| Total body cholesterol | Whole-body cholesterol mass across all tissues and fluids | g |
| Model-estimated transport floor | Calculated minimum plasma cholesterol concentration that satisfies the structural and delivery functions of the circulating lipoprotein system under stated assumptions | mg/dL |
| ■ Lipoprotein particle units are NMR conventions
NMR lipoprotein subfraction analysis (LipoProfile and similar) reports LDL-P in nanomolar (nmol/L), reflecting the ~10¹⁵–10¹⁸ particles/L range typical for adults. HDL-P is conventionally reported in micromolar (µmol/L) because HDL is more abundant on a per-particle basis. This paper uses the NMR conventions throughout and converts to particles per dL with explicit arithmetic. |
| ■ Plasma vs serum
Routine clinical lipid panels are performed on either serum or plasma; the difference in measured cholesterol is small (typically < 3%). Unless specified, all circulating cholesterol concentrations refer to plasma or serum interchangeably. |
2. Systemic Distribution and Mass Balance
2.1 Total body cholesterol
Total body cholesterol in an adult is approximately 120–150 g, with body-weight scaling. The range reflects between-subject and between-method variability in tissue-distribution studies. [2][3] Older textbook citations of ~35 g represent only Goodman’s rapidly-exchanging Pool A (liver + plasma + erythrocytes + part of viscera) in the kinetic tracer model, not whole-body content. Direct quantitative tissue analysis (Sabine, 1977, recapitulated in modern reviews) places adult human total body cholesterol at the higher figure, with brain, connective tissue including adipose, and skeletal muscle each contributing roughly 25–35 g. [2][3]
Goodman’s three-pool tracer model defines a rapidly-exchanging Pool A (liver, plasma, erythrocytes, splanchnic) of 15–30 g, a slowly-exchanging Pool B (skeletal muscle, adipose, dense connective tissue) of 35–60 g, and a kinetically isolated CNS pool of ~30–35 g. [4][5] The plasma compartment alone contains 3–7 g of cholesterol, depending on plasma volume and measured plasma total cholesterol concentration.
2.2 Tissue distribution
Distribution under the assumption of ~140 g total body cholesterol (representative midpoint). All entries are tissue cholesterol mass. [2][3]
| Compartment | Cholesterol (g) | % of body total | Dominant chemical form |
| Brain / CNS (myelin + neural membranes) | 30–35 | ~22–25% | >99.5% free (unesterified) |
| Skeletal muscle | ~25–30 | ~18–22% | Free (plasma membrane) |
| Connective tissue, adipose, body fluids | ~25–32 | ~18–23% | Mixed; CE accumulates with age in tendon/dura |
| Skin | ~12–18 | ~9–13% | Free |
| Other viscera (lung, kidney, intestine, etc.) | ~12–18 | ~9–13% | Free predominantly |
| Liver | ~4–6 | ~3–4% | Mixed FC + CE |
| Plasma / circulating pool | 3–7 | ~2–5% | ~70% CE / ~30% FC |
| Heart, spleen, adrenals (per organ) | <1 each | <1% | High density per gram (adrenals ≥ 25 mg/g) |
The CNS contains ~22–25% of body cholesterol at the highest tissue concentration (~23 mg/g wet weight). [5][6] The blood–brain barrier is impermeable to lipoprotein cholesterol; the entire CNS pool is synthesized locally. Cortical neuronal cholesterol turns over with a half-life of 6 months to 5 years; myelin cholesterol turns over on the order of decades. Net efflux from the CNS occurs primarily as 24S-hydroxycholesterol, which traverses the blood–brain barrier and is cleared in plasma. [5][6][7]
2.3 Plasma cholesterol mass at different plasma total cholesterol concentrations
Plasma cholesterol mass is the product of plasma volume and plasma total cholesterol concentration. At a representative plasma volume of 3.0 L:
| Plasma TC (mg/dL) | Plasma cholesterol mass (g, 3.0 L plasma) | Clinical context |
| 50 | 1.5 | Below typical neonatal range; rare LOF mutations |
| 80 | 2.4 | Reported PCSK9 LOF homozygote range |
| 100 | 3.0 | PCSK9 inhibitor on-treatment targets |
| 150 | 4.5 | Therapeutic target on intensive lipid-lowering |
| 188 | 5.6 | US adult mean (NHANES 2017–2018) [8] |
| 220 | 6.6 | Borderline-elevated |
| 280 | 8.4 | Heterozygous FH range |
| ■ Plasma volume varies with body size
Plasma volume in adults is approximately 40–45 mL/kg body weight (~2.8–3.2 L for a 70-kg adult, ~3.5–4.0 L for a 90-kg adult). All plasma mass calculations in this paper use a representative 3.0 L unless otherwise stated. |
2.4 Subcellular distribution and the ER cholesterol sensor
Within most non-neural cells, the plasma membrane holds 60–90% of cellular cholesterol at 30–40 mol% of PM lipids, while the endoplasmic reticulum maintains only 3–6 mol%. [9][10] The gradient is enforced by SREBP2 coupled to SCAP and Insig: when ER cholesterol exceeds ~5 mol%, SCAP retains SREBP2 in the ER and synthesis is suppressed. [11] Das et al. (eLife 2014) resolved PM cholesterol into three functional pools using a Perfringolysin O (PFO*) probe: a sphingomyelin-sequestered pool (~15 mol% of PM lipids), an “essential” pool required for cell viability, and an “accessible” pool that fluxes to the ER to regulate synthesis. [11][12] The accessible pool—not bulk PM cholesterol—is the regulatory signal.
| ▲ PFO* binding threshold applies to plasma-membrane bilayers, not lipoprotein monolayers.
The 35 mol% PFO*-accessibility threshold in plasma membranes reflects bilayer architecture and sphingomyelin–cholesterol complexation specific to the PM. [11][12] LDL and HDL surfaces are phospholipid monolayers stabilized by apolipoproteins; they have different lipid composition, leaflet asymmetry, and packing constraints. The threshold should not be applied directly to lipoprotein surfaces without further analysis. |
2.5 Daily turnover
Whole-body de novo cholesterol synthesis in adults is approximately 10 mg/kg/day (~700 mg/day in a 70-kg adult, ~880 mg/day in an 88-kg adult). The liver contributes only ~10% of total synthesis in humans; the majority is extrahepatic. [13] Three-pool tracer studies of plasma cholesterol turnover give a total production rate (synthesis plus dietary absorption) of ~1.0–1.2 g/day in normolipidemic subjects. [4] Dietary intake (~300–500 mg/day on a Western diet, ~30–50% absorbed) is compensated by down-regulation of endogenous synthesis. [14]
3. Lipoprotein Architecture and Cholesterol Partitioning
3.1 Representative 22-nm LDL particle composition
The reconstruction below is from Hevonoja et al. (BBA 2000) for a representative LDL particle of 22 nm diameter. [15][16][17]
| Component | Molecules / particle | Mass fraction (%) | Location |
| Cholesteryl esters (CE) | ~1,600 | 40–45% | Core |
| Unesterified cholesterol (FC) | ~600 | 8–10% | ~400 surface, ~200 core |
| Triglycerides (TG) | ~170 | 5–9% | Core |
| Phosphatidylcholine | ~450 | } 19–21% combined | Surface monolayer |
| Sphingomyelin | ~185 | (phospholipid total) | Surface monolayer |
| ApoB-100 (single copy) | 1 | 20–25% | Wraps the surface |
Total cholesterol per representative LDL particle is ~2,200 molecules; surface FC accounts for ~18% of LDL cholesterol. Some general references cite ~1,500 cholesterol per average LDL particle, reflecting different averaging schemes across the LDL size distribution; both numbers are central-tendency descriptions of an inherently heterogeneous population. [16]
| ▲ LDL is heterogeneous.
In-vivo LDL spans ~22–27.5 nm with substantial compositional variation; small dense LDL carries less cholesterol per particle and is enriched in TG, while large buoyant LDL carries more. The Hevonoja reconstruction is a representative central-tendency particle and should not be applied uncritically to all LDL species. |
3.2 Plasma free-to-esterified ratio
Direct enzymatic and chromatographic measurement in healthy human serum shows plasma free cholesterol at ~25–30% of total plasma cholesterol, with the esterified fraction at ~70–75%. [18][19] An elevated FC/CE ratio is associated with LCAT dysfunction, familial chylomicronemia, and an independent atherogenic signal in some studies. [20]
3.3 Surface free cholesterol: structural role
Free cholesterol on the lipoprotein surface intercalates between phospholipid acyl chains, with its 3β-hydroxyl projecting into the aqueous interface and its rigid sterol ring aligned with the lipid tails. This “condensation” reduces free volume in the monolayer, decreases its permeability to water, and increases mechanical stability of the particle. [21][22] FC at ~25 mol% of surface lipids is compatible with a liquid-ordered, well-packed monolayer; this configuration supports particle stability and lateral mobility for enzymatic processing. [23]
3.4 Free cholesterol exchange
Free cholesterol on lipoprotein surfaces is in spontaneous equilibrium with cell membranes and other lipoprotein particles via passive aqueous diffusion. Exchange half-time scales inversely with surface curvature: ~5 min for nascent HDL, ~45 min for LDL. [21][23] This kinetic system maintains a near-uniform thermodynamic activity of cholesterol across all extracellular lipid surfaces. [22][23]
4. Cholesteryl Esters: Transport Cargo
Cholesteryl esters are fully hydrophobic and reside in the lipoprotein core as a liquid or liquid-crystalline droplet. CE in LDL represents 40–45% of particle mass and provides the bulk of cholesterol delivered to cells via receptor-mediated endocytosis, where lysosomal acid lipase liberates free cholesterol for use. [17]
CETP shuttles CE from HDL to apoB-containing lipoproteins in exchange for triglycerides, enriching apoB particles with cholesterol cargo. A substantial fraction of apoB-particle core CE therefore reflects cholesterol that has cycled through peripheral cells, plasma, and HDL before being transferred onto LDL, superimposed on hepatic cholesterol pools secreted as VLDL. [24]
5. HDL Maturation
Nascent HDL is secreted as lipid-poor apoA-I that acquires phospholipid and free cholesterol from peripheral cells via ABCA1, forming a discoidal particle of two apoA-I molecules in a “double-belt” conformation. [25] LCAT, activated by apoA-I, transfers an acyl group from the sn-2 position of phosphatidylcholine to surface FC, generating CE that migrates into the hydrophobic core. [23][25]
As CE accumulates, the discoidal bilayer becomes spherical. Mature HDL2 reaches ~10–12 nm diameter and carries ~30–60 cholesterol molecules per particle (mostly CE), with ~5–15 surface FC. [23] The CE either returns directly to the liver via SR-B1 or transfers to apoB particles via CETP for hepatic clearance through LDLR. [23][24]
6. Tissue Requirements: De Novo Synthesis and Exogenous Uptake
6.1 Most nucleated tissues can synthesize cholesterol de novo
Every nucleated cell in the human body expresses the complete mevalonate–cholesterol biosynthetic pathway. [13][26] Under usual conditions, most tissues are not absolutely dependent on continuous exogenous cholesterol supply from plasma; the quantitative balance between local synthesis and lipoprotein-derived uptake varies by tissue, age, and physiological state.
| Tissue | Primary cholesterol source | Notes |
| Brain / CNS | Local synthesis (astrocytes → neurons via apoE) | BBB excludes lipoprotein cholesterol; net efflux via 24S-OH-cholesterol [7] |
| Skeletal muscle | Predominantly local synthesis | Low demand for new sterol |
| Skin | ~90% local synthesis | Supports barrier function |
| Adrenal cortex | Mixed: LDLR > SR-B1 in humans; reverse in rodents [27][28] | Stored CE buffers acute steroidogenic demand |
| Gonads (testes, ovaries) | Mixed: LDLR + SR-B1; reproductive-state-dependent | Pregnancy and lactation raise demand |
| Placenta | Maternal LDL and HDL; pregnancy only | Outside scope of nonpregnant adult analysis |
| Liver | Synthesis + dietary + reverse transport | Master regulator; secretes VLDL |
In human adrenocortical and gonadal physiology, LDLR-mediated endocytosis carries greater quantitative weight than SR-B1 for cholesterol delivery — opposite to the rodent pattern in which SR-B1 dominates. SR-B1 nonetheless remains expressed and biologically active in human steroidogenic tissues, and SR-B1 loss-of-function in humans produces subtle but real abnormalities in ACTH-stimulated cortisol response. The mixed-pathway nature of human steroidogenic cholesterol supply should be preserved in any mechanistic discussion. [27][28]
6.2 LDL-receptor kinetics: in-vitro and in-vivo are not interchangeable
In-vitro: in cultured human fibroblasts, high-affinity binding of LDL apoB-100 saturates at LDL protein concentrations below 50 µg/mL (Brown & Goldstein). [29][30] Catapano and colleagues (2024) describe this as a half-saturation corresponding to an LDL-C plasma equivalent of ~2.5 mg/dL, and combine it with the observation that interstitial-fluid LDL-C is ~20% of plasma LDL-C to argue that plasma LDL-C of ~12.5 mg/dL would saturate tissue LDLR. [31] This argument depends on intermediate assumptions about LDL protein-to-cholesterol mass ratio, interstitial-fluid composition, and uniformity of fibroblast LDLR behavior; this paper cites the argument without endorsing it as a measured human threshold.
In-vivo: organ-level LDL clearance in rat and hamster (Spady & Dietschy) gives Km ≈ 90 mg/dL — roughly 30-fold higher, reflecting unstirred boundary layers, capillary permeability, and receptor density rather than intrinsic affinity. [32] This rodent in-vivo number is not directly portable to human tissue-level cholesterol-delivery thresholds.
| ▲ Receptor saturation kinetics are not a clean human delivery threshold.
The chain (in-vitro fibroblast Km → plasma-equivalent LDL-C → interstitial-fluid LDLR saturation in adrenal/gonadal cells) depends on assumptions that have not been independently validated in humans, and hepatocytes are exposed to sinusoidal blood rather than to ordinary interstitial fluid. The substantively defensible claim is empirical: humans with very low plasma LDL-C, by genetics or therapy, have not shown clinical signs of cholesterol-delivery insufficiency in the reported populations. |
6.3 Empirical observations: very low LDL-C in humans
Carriers of homozygous PCSK9 loss-of-function mutations are a small reported sample. The originally described Dallas Heart Study African-American homozygotes had lifelong plasma LDL-C of 14–29 mg/dL (total cholesterol typically 50–80 mg/dL) without overt clinical abnormalities in the limited published descriptions. [33][34] The total reported global population of such homozygotes remains small, so this evidence supports the absence of large-effect harm but does not prove a universal physiological minimum.
Larger evidence comes from PCSK9-inhibitor randomized trials. FOURIER (n = 27,564) had a median randomized follow-up of 2.2 years, with on-treatment median LDL-C of 30 mg/dL and no signal of adrenal insufficiency, hypogonadism, cognitive decline, hemorrhagic stroke, or muscle disease. [35] ODYSSEY OUTCOMES (n = 18,924) had a median follow-up of 2.8 years with similar safety findings. [36] The FOURIER Open-Label Extension (FOURIER-OLE, n = 6,635 of the original 27,564) added a median 5.0 years of evolocumab exposure, bringing cumulative exposure in that subset to ~8.4 years, with no new safety signal. [37] These are large, prospective safety data; they are not a direct measurement of human cholesterol delivery thresholds but they bound the empirically demonstrated safe range of plasma LDL-C from above.
Interstitial-fluid LDL is reported at ~10–20% of plasma LDL concentration in human peripheral lymph studies. [38][39] These data are descriptive and do not establish a measured human tissue saturation threshold.
7. A Transport-Model Estimate of the Minimum Circulating Cholesterol
| ■ What this section is and is not.
This section presents a transport-model estimate of the minimum plasma cholesterol concentration that simultaneously (i) sustains the structural integrity of the circulating lipoprotein system and (ii) supplies the net daily exogenous cholesterol demand of a nonpregnant adult under usual conditions. The estimate is calculation, not measurement. Equations and parameter values are in Section 11. |
7.1 Structural integrity
All circulating lipoprotein particles require surface free cholesterol to stabilize their phospholipid monolayer. Using NMR-consistent particle counts (LDL-P in nmol/L; HDL-P in µmol/L) and explicit unit conversion: [15][17]
| Particle class | Class concentration (clinical) | Particles per dL | Surface FC per particle | Surface FC contribution (mg/dL) |
| LDL | LDL-C 100 mg/dL; LDL-P ~1,200 nmol/L | ~7.1 × 10¹⁶ | ~400 | ~18 |
| HDL | HDL-C 50 mg/dL; HDL-P ~30 µmol/L | ~1.8 × 10¹⁸ | ~10 | ~12 |
| VLDL / IDL | TG-driven; particle counts ~10¹⁵–10¹⁶ /dL | — | ~variable | ~2–3 |
| Total surface FC (model, baseline TC ~190) | — | — | — | ~30–33 |
| Total measured plasma FC (~28% of TC at TC 190) | — | — | — | ~50–55 (includes core FC and other) |
The model-derived surface FC of ~30–33 mg/dL at typical clinical lipid values, plus core FC of ~10–15 mg/dL (LDL has ~200 core FC per particle, HDL has a smaller core FC contribution), and a small chylomicron-remnant and Lp(a) contribution, sums to approximately 45–55 mg/dL plasma FC. This matches the directly measured plasma FC fraction of ~25–30% of total cholesterol at total cholesterol ~190 mg/dL. [18][19]
7.2 Net daily exogenous cholesterol delivery to peripheral tissues
Reverse cholesterol transport tracer studies in healthy adults (Turner et al., 2012) estimate whole-body tissue FC efflux at 3.79 ± 0.88 mg/kg/h, reported in the original paper as ≈8 g/day; this corresponds to ~6.4 g/day at 70 kg or ~8 g/day at 88 kg (the study’s mean approximate body weight). [40] This is bidirectional exchange flux, not net delivery demand. Estimated net obligate exogenous uptake by tissues whose local synthesis is insufficient under baseline conditions, in a nonpregnant adult, is approximately:
| Tissue / function | Estimated daily cholesterol use | Source of supply |
| Adrenal steroidogenesis (baseline) | ~30–60 mg/day | Mostly LDLR-mediated; SR-B1 contribution |
| Gonadal steroidogenesis (nonpregnant adult, baseline) | < 10 mg/day | Predominantly LDLR-mediated in humans |
| Bile acid synthesis | ~400 mg/day | Hepatic pool; not a peripheral demand |
| Extrahepatic membrane turnover (all) | ~600–700 mg/day | Met by local synthesis |
| Net obligate exogenous demand (nonpregnant adult, baseline) | Order of tens of mg/day (~50 mg/day central estimate) | Met at very low plasma LDL-C |
| ▲ Nonpregnant adult, baseline only.
Pregnancy roughly doubles maternal plasma cholesterol; placental demand is on a different order of magnitude and is not captured here. Acute illness, severe stress, and rapid tissue regeneration also raise demand. |
7.3 Combined model-estimated transport floor
Model output across a range of plasma TC values:
| Plasma TC scenario | LDL surface FC (mg/dL) | HDL surface FC (mg/dL) | Total plasma FC (mg/dL) | Notes |
| TC 188 (US mean) | ~18 | ~12 | ~50–55 | Baseline |
| TC 100 (PCSK9-Rx target) | ~10 | ~10 | ~30 | Adequate; well above any structural floor |
| TC 70 (PCSK9 LOF homozygote) | ~4 | ~9 | ~17 | At or near model floor; empirically tolerated |
| TC 50 (extreme genetic) | ~2 | ~8 | ~13 | Below any well-validated empirical case |
Bottom line: the transport model predicts that a nonpregnant adult under baseline conditions can maintain a functional circulating lipoprotein system and meet ~50 mg/day of exogenous tissue demand at plasma total cholesterol on the order of 50–90 mg/dL (sensitivity range 40–110 mg/dL). The chemical form remains roughly 70% esterified and 30% unesterified across this range. The model-floor estimate is in numerical agreement with the plasma TC observed in PCSK9 LOF homozygotes (~50–80 mg/dL), providing independent corroboration from clinical and genetic observation. Modern US adult mean plasma TC of ~188 mg/dL [8] exceeds the model-estimated floor by approximately 2–4×, depending on which sensitivity scenario is used.
7.4 Sensitivity bounds
| Assumption varied | Range tested | Effect on transport-floor estimate |
| Plasma volume | 2.5–4.0 L | Inverse scaling; primary effect on absolute mass, secondary on mg/dL |
| LDL-P at given LDL-C | ± 50% of representative | Affects FC component proportionally |
| Surface FC per LDL particle | 300–500 | ± 15% on FC component |
| FC per HDL particle | 5–15 | ± 25% on HDL FC component |
| Net peripheral demand | 20–150 mg/day | Adds 5–30 mg/dL margin |
| Pregnancy / acute illness | — | Model not applicable; demand can rise 2–10× |
| Combined plausible range (nonpregnant adult, baseline) | — | ~40–110 mg/dL plasma TC |
8. ApoB Causality and ASCVD Risk
Whether peripheral tissues require circulating cholesterol is a separate question from whether circulating apoB-containing lipoproteins are causally atherogenic. Mendelian randomization across > 50 genetic instruments (LDLR, PCSK9, HMGCR, NPC1L1, APOB, ANGPTL3, LPL, CETP) has established that the lifetime risk of ASCVD tracks cumulative apoB-particle exposure rather than absolute LDL-C concentration alone, with a log-linear relationship between apoB and event risk extending below 20 mg/dL LDL-C. [41][42]
The atherogenic risk imposed by a given circulating cholesterol mass is determined by particle number (apoB count) and residence time, not by whether the cholesterol is required for tissue delivery. The two questions are physiologically orthogonal.
9. Evolutionary and Mechanistic Context
Cholesterol biosynthesis requires 11 molecules of O₂ and roughly 100 ATP equivalents per cholesterol molecule, and involves toxic intermediates. [43][44] This expense is consistent with cholesterol having emerged as eukaryotes adapted to a rising atmospheric oxygen tension; cholesterol both consumes O₂ during synthesis and reduces membrane O₂ permeability, partially functioning as an O₂ sink. [44]
Under the modeling perspective developed in Section 7, the lipoprotein transport system is best interpreted as (i) a vehicle for triglyceride distribution — LDL is a kinetic byproduct of VLDL catabolism — and (ii) a kinetic chemical-potential buffer that maintains uniform sterol activity across all extracellular lipid surfaces. [22][23] Under this interpretation, the modern population’s circulating cholesterol concentration is set primarily by hepatic clearance capacity (LDLR density, PCSK9 activity, IDOL activity) rather than by peripheral demand.
10. Conclusions
- Total body cholesterol in an adult is approximately 120–150 g; the plasma compartment of 3–7 g represents 2–5%, not 10–30%, of body content.
- The brain contains ~30–35 g (22–25% of body cholesterol), entirely synthesized locally and isolated from plasma by the blood–brain barrier.
- A representative 22-nm LDL particle contains ~2,200 cholesterol molecules (~1,600 CE + ~600 FC, of which ~400 FC are on the surface). LDL is heterogeneous; these numbers describe a representative reconstruction.
- Plasma cholesterol is ~70% esterified and ~30% free.
- In humans, LDLR-mediated endocytosis carries greater weight than SR-B1 for adrenal and gonadal cholesterol uptake, but SR-B1 remains biologically active.
- Using NMR-consistent lipoprotein particle counts (LDL-P in nmol/L, HDL-P in µmol/L), the model-estimated minimum plasma total cholesterol for a nonpregnant adult under baseline conditions is approximately 50–90 mg/dL (sensitivity 40–110 mg/dL). This is a calculated scenario, not a directly measured human physiological minimum.
- Net daily exogenous cholesterol delivery to peripheral tissues in a nonpregnant adult under baseline conditions is on the order of tens of mg/day, easily met at plasma LDL-C in the 15–30 mg/dL range observed in PCSK9 LOF carriers and in PCSK9-inhibitor trial subjects (FOURIER median 2.2 years, FOURIER-OLE subset median 5.0 additional years), in whom no signal of cholesterol-delivery insufficiency has been reported.
- US adult mean plasma total cholesterol of ~188 mg/dL (NHANES 2017–2018) exceeds the model-estimated transport floor by approximately 2–4×, depending on which sensitivity scenario is used. The surplus is metabolically tolerated, causally tied to ASCVD risk on a per-apoB basis, and not required for any known physiological function in a nonpregnant adult under baseline conditions.
11. Methods Appendix
11.1 Scope
This appendix specifies the equations, parameter values, and assumptions used in the transport-model estimate of Section 7.
11.2 Equations
Plasma cholesterol mass:
M_plasma = TC × V_plasma / 1000 (g)
where TC is in mg/dL and V_plasma is in dL.
Conversion from NMR particle concentration to particles per dL:
N_particles_per_dL = [class-P] × 10⁻⁹ × N_A / 10 (LDL: [LDL-P] in nmol/L)
N_particles_per_dL = [class-P] × 10⁻⁶ × N_A / 10 (HDL: [HDL-P] in µmol/L)
where N_A = 6.022 × 10²³ /mol. The /10 converts L to dL.
Worked example, LDL-P = 1,200 nmol/L:
1,200 × 10⁻⁹ × 6.022 × 10²³ / 10 = 7.23 × 10¹⁶ particles/dL
Worked example, HDL-P = 30 µmol/L:
30 × 10⁻⁶ × 6.022 × 10²³ / 10 = 1.81 × 10¹⁸ particles/dL
Surface FC contribution from one lipoprotein class:
[FC]_class (mg/dL) = N_particles_per_dL × n_FC,class × MW_chol × 1000 / N_A
Worked example, LDL at LDL-C 100 (N = 7.23 × 10¹⁶/dL), 400 surface FC per particle, MW_chol = 386.7 g/mol:
7.23 × 10¹⁶ × 400 × 386.7 × 1000 / 6.022 × 10²³ = ~18.2 mg/dL
Total model-estimated transport floor:
TC_floor ≈ Σ [FC]_class,surface + Σ [FC]_class,core + CE_core,structural + CE_delivery_reserve
11.3 Parameter values used
| Parameter | Central value | Source / basis | Sensitivity range |
| Plasma volume | 3.0 L (= 30 dL) | ~42 mL/kg × 70 kg | 2.5–4.0 L |
| LDL-P at LDL-C 100 mg/dL | ~1,200 nmol/L (= 7.1 × 10¹⁶ /dL) | NMR LipoProfile data; MESA, JUPITER | 600–1,800 nmol/L |
| HDL-P at HDL-C 50 mg/dL | ~30 µmol/L (= 1.8 × 10¹⁸ /dL) | NMR data | 20–40 µmol/L |
| Surface FC per LDL particle | 400 | Hevonoja 2000 reconstruction [15] | 300–500 |
| FC per HDL particle (total) | ~10 | HDL2/3 composition reviews; calculated from HDL-C / HDL-P at central values | 5–15 |
| Net daily peripheral exogenous demand (nonpregnant adult) | ~50 mg/day | Composite of adrenal/gonadal/membrane estimates [26][27] | 20–150 mg/day |
| MW cholesterol | 386.7 g/mol | Fixed | n/a |
| Avogadro’s number | 6.022 × 10²³ /mol | Fixed | n/a |
11.4 Assumptions
The transport-floor calculation depends on assumptions made explicit here:
- The nonpregnant adult is at metabolic steady state under baseline conditions (no pregnancy, no acute illness, no rapid tissue regeneration).
- Plasma volume is taken as a single representative value rather than individualized.
- Particle counts scale approximately linearly with class cholesterol concentration over the modeled range; this approximation breaks down at extremes.
- Surface FC per LDL is taken from the Hevonoja reconstruction; surface composition varies somewhat with particle size and remodeling state.
- FC per HDL particle reflects mature spherical HDL2/3 composition; nascent discoidal HDL has different per-particle FC content.
- Net peripheral demand is taken as order of tens of mg/day; this aggregates tissue-level values that have wide uncertainty under different endocrine states.
- Lp(a), chylomicrons, and chylomicron remnants are not modeled separately; including them would shift the FC contribution upward by a small amount.
- The model is steady-state and ignores diurnal, postprandial, and seasonal variation.
11.5 What this appendix does not establish
This appendix does not establish a measured human physiological minimum for total cholesterol. It provides a transparent, reproducible transport-model estimate. Direct empirical evidence for the safety of very low plasma LDL-C in humans (PCSK9 LOF carriers; PCSK9-inhibitor trial participants) is presented separately in Section 6.3 and remains the strongest evidence that the model’s predicted low floor is physiologically plausible. The two evidence streams are independent: a transport model and a clinical/genetic observation. They are numerically consistent — both indicate that plasma total cholesterol can fall to ~50–80 mg/dL without identified clinical consequence in a nonpregnant adult — but neither alone is dispositive.
References
- Mach F, Baigent C, Catapano AL, et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. 2020;41(1):111-188. doi:10.1093/eurheartj/ehz455
- Pikuleva IA, Curcio CA. Cholesterol in the retina: the best is yet to come. Prog Retin Eye Res. 2014;41:64-89. doi:10.1016/j.preteyeres.2014.03.002
- Mc Auley MT, Wilkinson DJ, Jones JJ, Kirkwood TB. A whole-body mathematical model of cholesterol metabolism and its age-associated dysregulation. BMC Syst Biol. 2012;6:130. Published 2012 Oct 10. doi:10.1186/1752-0509-6-130
- Goodman DS, Noble RP, Dell RB. Three-pool model of the long-term turnover of plasma cholesterol in man. J Lipid Res. 1973;14(2):178-188.
- Dietschy JM, Turley SD. Thematic review series: brain Lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J Lipid Res. 2004;45(8):1375-1397. doi:10.1194/jlr.R400004-JLR200
- Björkhem I, Meaney S. Brain cholesterol: long secret life behind a barrier. Arterioscler Thromb Vasc Biol. 2004;24(5):806-815. doi:10.1161/01.ATV.0000120374.59826.1b
- Lütjohann D, Breuer O, Ahlborg G, et al. Cholesterol homeostasis in human brain: evidence for an age-dependent flux of 24S-hydroxycholesterol from the brain into the circulation. Proc Natl Acad Sci U S A. 1996;93(18):9799-9804. doi:10.1073/pnas.93.18.9799
- Gao Y, Shah LM, Ding J, Martin SS. US Trends in Cholesterol Screening, Lipid Levels, and Lipid-Lowering Medication Use in US Adults, 1999 to 2018. J Am Heart Assoc. 2023;12(3):e028205. doi:10.1161/JAHA.122.028205
- Canfrán-Duque A, Rotllan N, Zhang X, et al. Macrophage-Derived 25-Hydroxycholesterol Promotes Vascular Inflammation, Atherogenesis, and Lesion Remodeling. Circulation. 2023;147(5):388-408. doi:10.1161/CIRCULATIONAHA.122.059062
- van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol. 2008;9(2):112-124. doi:10.1038/nrm2330
- Das A, Brown MS, Anderson DD, Goldstein JL, Radhakrishnan A. Three pools of plasma membrane cholesterol and their relation to cholesterol homeostasis. Elife. 2014;3:e02882. Published 2014 Jun 11. doi:10.7554/eLife.02882
- Schoop V, Martello A, Eden ER, Höglinger D. Cellular cholesterol and how to find it. Biochim Biophys Acta Mol Cell Biol Lipids. 2021;1866(9):158989. doi:10.1016/j.bbalip.2021.158989
- Dietschy JM, Turley SD, Spady DK. Role of liver in the maintenance of cholesterol and low density lipoprotein homeostasis in different animal species, including humans. J Lipid Res. 1993;34(10):1637-1659.
- Jones PJ, Pappu AS, Hatcher L, Li ZC, Illingworth DR, Connor WE. Dietary cholesterol feeding suppresses human cholesterol synthesis measured by deuterium incorporation and urinary mevalonic acid levels. Arterioscler Thromb Vasc Biol. 1996;16(10):1222-1228. doi:10.1161/01.atv.16.10.1222
- Hevonoja T, Pentikäinen MO, Hyvönen MT, Kovanen PT, Ala-Korpela M. Structure of low density lipoprotein (LDL) particles: basis for understanding molecular changes in modified LDL. Biochim Biophys Acta. 2000;1488(3):189-210. doi:10.1016/s1388-1981(00)00123-2
- Kumar V, Butcher SJ, Öörni K, et al. Three-dimensional cryoEM reconstruction of native LDL particles to 16Å resolution at physiological body temperature. PLoS One. 2011;6(5):e18841. Published 2011 May 9. doi:10.1371/journal.pone.0018841
- Feingold KR. Introduction to Lipids and Lipoproteins. In: Feingold KR, Adler RA, Ahmed SF, et al., eds. Endotext. South Dartmouth (MA): MDText.com, Inc.; January 14, 2024.
- Leonard PJ, Shaper AG, Jones KW. Relationship between free and total cholesterol values in human serum. Am J Clin Nutr. 1965;17(6):377-380. doi:10.1093/ajcn/17.6.377
- Saier EL, Nordstrand E, Juves MW, Hartsock RJ. The ratio of free to esterified cholesterol in serum. A new discriminant in correlating lipid metabolism with disease state. Am J Clin Pathol. 1979;71(1):83-87. doi:10.1093/ajcp/71.1.83
- Bagheri B, Alikhani A, Mokhtari H, Rasouli M. The Ratio of Unesterified/esterified Cholesterol is the Major Determinant of Atherogenicity of Lipoprotein Fractions. Med Arch. 2018;72(2):103-107. doi:10.5455/medarh.2018.72.103-107
- Lange Y, Steck TL. Cholesterol homeostasis and the escape tendency (activity) of plasma membrane cholesterol. Prog Lipid Res. 2008;47(5):319-332. doi:10.1016/j.plipres.2008.03.001
- Maxfield FR, Tabas I. Role of cholesterol and lipid organization in disease. Nature. 2005;438(7068):612-621. doi:10.1038/nature04399
- Rothblat GH, Phillips MC. High-density lipoprotein heterogeneity and function in reverse cholesterol transport. Curr Opin Lipidol. 2010;21(3):229-238. doi:10.1097/mol.0b013e328338472d
- Barter PJ, Brewer HB Jr, Chapman MJ, Hennekens CH, Rader DJ, Tall AR. Cholesteryl ester transfer protein: a novel target for raising HDL and inhibiting atherosclerosis. Arterioscler Thromb Vasc Biol. 2003;23(2):160-167. doi:10.1161/01.atv.0000054658.91146.64
- Shih AY, Sligar SG, Schulten K. Maturation of high-density lipoproteins. J R Soc Interface. 2009;6(39):863-871. doi:10.1098/rsif.2009.0173
- Cerqueira NM, Oliveira EF, Gesto DS, et al. Cholesterol Biosynthesis: A Mechanistic Overview. Biochemistry. 2016;55(39):5483-5506. doi:10.1021/acs.biochem.6b00342
- Azhar S, Reaven E. Scavenger receptor class BI and selective cholesteryl ester uptake: partners in the regulation of steroidogenesis. Mol Cell Endocrinol. 2002;195(1-2):1-26. doi:10.1016/s0303-7207(02)00222-8
- Gomez-Sanchez CE, Gomez-Sanchez EP. Cholesterol Availability and Adrenal Steroidogenesis. Endocrinology. 2024;165(4):bqae032. doi:10.1210/endocr/bqae032
- Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986;232(4746):34-47. doi:10.1126/science.3513311
- Goldstein JL, Brown MS. Familial hypercholesterolemia: identification of a defect in the regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity associated with overproduction of cholesterol. Proc Natl Acad Sci U S A. 1973;70(10):2804-2808. doi:10.1073/pnas.70.10.2804
- Borghi C, Bragagni A. There are those who would like zero LDL cholesterol. Eur Heart J Suppl. 2024;26(Suppl 1):i19-i22. Published 2024 Apr 17. doi:10.1093/eurheartjsupp/suae012
- Spady DK, Bilheimer DW, Dietschy JM. Rates of receptor-dependent and -independent low density lipoprotein uptake in the hamster. Proc Natl Acad Sci U S A. 1983;80(11):3499-3503. doi:10.1073/pnas.80.11.3499
- Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354(12):1264-1272. doi:10.1056/NEJMoa054013
- Zhao Z, Tuakli-Wosornu Y, Lagace TA, et al. Molecular characterization of loss-of-function mutations in PCSK9 and identification of a compound heterozygote. Am J Hum Genet. 2006;79(3):514-523. doi:10.1086/507488
- Sabatine MS, Giugliano RP, Keech AC, et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N Engl J Med. 2017;376(18):1713-1722. doi:10.1056/NEJMoa1615664
- Schwartz GG, Steg PG, Szarek M, et al. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N Engl J Med. 2018;379(22):2097-2107. doi:10.1056/NEJMoa1801174
- O’Donoghue ML, Giugliano RP, Wiviott SD, et al. Long-Term Evolocumab in Patients With Established Atherosclerotic Cardiovascular Disease. Circulation. 2022;146(15):1109-1119. doi:10.1161/CIRCULATIONAHA.122.061620
- Sloop CH, Dory L, Roheim PS. Interstitial fluid lipoproteins. J Lipid Res. 1987;28(3):225-237.
- Reichl D, Simons LA, Myant NB, Pflug JJ, Mills GL. The lipids and lipoproteins of human peripheral lymph, with observations on the transport of cholesterol from plasma and tissues into lymph. Clin Sci Mol Med. 1973;45(3):313-329. doi:10.1042/cs0450313
- Turner S, Voogt J, Davidson M, et al. Measurement of reverse cholesterol transport pathways in humans: in vivo rates of free cholesterol efflux, esterification, and excretion. J Am Heart Assoc. 2012;1(4):e001826. doi:10.1161/JAHA.112.001826
- Ference BA, Ginsberg HN, Graham I, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2017;38(32):2459-2472. doi:10.1093/eurheartj/ehx144
- Sniderman AD, Thanassoulis G, Glavinovic T, et al. Apolipoprotein B Particles and Cardiovascular Disease: A Narrative Review. JAMA Cardiol. 2019;4(12):1287-1295. doi:10.1001/jamacardio.2019.3780
- Summons RE, Bradley AS, Jahnke LL, Waldbauer JR. Steroids, triterpenoids and molecular oxygen. Philos Trans R Soc Lond B Biol Sci. 2006;361(1470):951-968. doi:10.1098/rstb.2006.1837
- Brown AJ, Galea AM. Cholesterol as an evolutionary response to living with oxygen. Evolution. 2010;64(7):2179-2183. doi:10.1111/j.1558-5646.2010.01011.x