Apolipoprotein B-Driven Atherosclerosis as a Causal Basis for Alzheimer’s Disease
A Multidisciplinary Perspective on the Cardiorenal-Cerebral Axis
Abstract
Background. The dominant neurocentric framework defines Alzheimer’s disease (AD) biologically by amyloid and tau biomarkers and treats vascular pathology as a frequent but secondary modifier. Genetic, epidemiological, and imaging evidence increasingly support an alternative reading in which lifelong exposure to apolipoprotein B (ApoB)-containing lipoproteins is a causal upstream driver of the cerebrovascular and small-vessel injury that, in a substantial subset of patients, precedes and accelerates AD-defining pathology.
Objective. To synthesize current evidence linking lifelong ApoB exposure to atherosclerotic injury across the carotid, intracranial, and renal vascular beds and, in turn, to the cognitive syndrome clinically diagnosed as Alzheimer’s disease.
Methods. Structured narrative review of PubMed-indexed primary studies, Mendelian randomization analyses, longitudinal imaging cohorts, randomized cardiovascular and dementia-prevention trials, and current consensus statements from the American Heart Association, the National Lipid Association, and the Alzheimer’s Association. Causal claims were evaluated against STROBE-MR reporting standards.[29]
Conclusions. Lifelong ApoB-containing lipoprotein exposure is causally established as a driver of atherosclerotic vascular disease. Converging evidence from drug-target Mendelian randomization in over one million individuals,[7] longitudinal PET imaging of intracranial stenosis,[13] and randomized vascular-risk-reduction trials[14] supports a causal contribution of this vascular injury to cognitive decline, to vascular cognitive impairment and dementia (VCID), and to the mixed-pathology dementia phenotypes that dominate older-adult autopsy series.[2,32] The extension from cerebrovascular injury to biomarker-confirmed Alzheimer’s disease as defined under the 2024 NIA-AA revised criteria[40] is supported but with appropriately weaker confidence, and the language of this manuscript is hedged accordingly at that endpoint. The 2024 NIA-AA criteria are a diagnostic framework, not an etiologic claim; they are fully compatible with both an established independent pathogenic role for amyloid-β (most directly demonstrated by autosomal-dominant familial AD)[41] and a causal upstream role for ApoB-driven cerebrovascular injury in sporadic late-onset disease. Lifelong ApoB reduction — through high-quality plant-forward dietary patterns and, where indicated, pharmacotherapy initiated in midlife — is a defensible prevention strategy for cognitive decline and a plausible component of dementia and AD prevention.
Introduction
The prevailing neurocentric model of Alzheimer’s disease, which prioritizes the amyloid-β cascade as the primary initiator of neurodegeneration, is increasingly challenged by genetic, epidemiological, and imaging evidence indicating that vascular health is not merely a comorbid factor but a foundational upstream driver of the disease process.[1,5,17] This analysis investigates the hypothesis that lifetime exposure to elevated apolipoprotein B (ApoB)-containing lipoproteins drives progressive atherosclerotic plaque accumulation in the carotid, intracranial, and renal arteries, initiating a cascade of systemic and local injuries that culminate in dementia in a substantial subset of patients.[4,7,20,22] By examining overlapping mechanisms of plaque formation across vascular beds and the causal inferences afforded by Mendelian randomization, a rigorous framework emerges for understanding Alzheimer’s disease as the late-stage neurological manifestation of decades-long vascular-lipid-inflammatory injury.[2,3,9]
Causal language is used deliberately but is calibrated to the strength of evidence at each link in the chain. ApoB is established as causal for atherosclerotic cardiovascular disease by triangulation across multivariable Mendelian randomization,[21,25] cumulative-exposure analyses,[22,33] and randomized lipid-lowering trials.[26] The extension of this causal pathway to vascular cognitive impairment and to mixed-pathology dementia is well-supported by drug-target Mendelian randomization for non-HDL-C and dementia in over one million individuals[7] and by longitudinal PET imaging linking baseline intracranial stenosis to subsequent amyloid and tau accumulation.[13] The further extension to biomarker-confirmed Alzheimer’s disease specifically — AD as defined by the 2024 NIA-AA revised criteria[40] — is supported by directionally consistent ApoB-specific Mendelian randomization[4] and by the mechanistic compatibility of vascular injury with amyloid and tau biology,[16,17,42] but at appropriately weaker strength than the cardiovascular link. The manuscript adopts hedged language at the AD endpoint accordingly and explicitly distinguishes all-cause dementia, vascular cognitive impairment, mixed-pathology dementia, and biomarker-confirmed AD throughout.
The Vascular-Atherosclerotic Hypothesis of Alzheimer’s Disease and Dementia
The vascular-atherosclerotic hypothesis posits that the clinical syndrome of Alzheimer’s disease is, in a substantial subset of patients, the downstream consequence of chronic cerebral hypoperfusion and neurovascular unit dysfunction initiated by large-artery atherosclerosis and propagated through the cerebral small-vessel network.[1,5,17] In this framework, focal narrowing of carotid and intracranial arteries, combined with loss of arterial elasticity, creates a hemodynamic environment hostile to neuronal survival and protein clearance.[12,18]
Contemporary neuropathological evidence demonstrates that the classical categorical distinction between pure vascular dementia and pure Alzheimer’s disease is empirically untenable in older adults.[3,32] Community-based autopsy studies report that mixed neuropathologies — any combination of Alzheimer-type, Lewy body, TDP-43, and vascular lesions — are present in the majority of aged brains, and pure proteinopathy without cerebrovascular pathology is the exception rather than the rule.[3,32] Large-vessel atherosclerosis, including carotid stenosis and intracranial atherosclerotic disease (ICAD), commonly coexists with cerebral small-vessel disease characterized by white-matter hyperintensities, lacunar infarcts, and microbleeds.[3,8,9]
Arterial stiffness — particularly in the proximal aorta and carotid arteries — transmits high-pressure pulsatility into the delicate microvasculature of the brain.[12] This hyperpulsatile flow damages the blood-brain barrier and disrupts the glymphatic system, a perivascular network responsible for clearing metabolic waste, including amyloid-β and tau.[15,18] In this framework, accumulation of these proteins is, in part, a consequence of impaired clearance rather than solely a consequence of overproduction.[16,17,18]
This hypothesis is not exclusive of canonical AD genetic risk. The apolipoprotein E ε4 (APOE ε4) allele — the largest common genetic risk factor for late-onset AD — operates substantially through cerebrovascular pathways. Direct human imaging evidence demonstrates that APOE ε4 carriers exhibit blood-brain barrier breakdown that precedes and predicts cognitive decline independently of amyloid and tau,[42] providing a mechanistic bridge between systemic lipoprotein biology, cerebrovascular injury, and the clinical AD phenotype.[5,16,42]
We engage explicitly with the 2024 Alzheimer’s Association revised criteria,[40] which define AD biologically by core amyloid and tau biomarkers, independent of clinical syndrome. These criteria are a diagnostic framework, not an etiologic claim, and the working group recognizes vascular and other co-pathologies as common modifiers of disease course. Our position is therefore not that the 2024 criteria are wrong or that they deny vascular contributions — they do neither. Our position is that the biomarker fingerprint they operationalize has, for a substantial subset of patients, an upstream cause: decades of ApoB-driven cerebrovascular injury interacting with intrinsic amyloid and tau biology. Defining AD by its downstream biomarker fingerprint is fully compatible with attributing that fingerprint, in part, to upstream vascular causes.
Table 1. Overlapping Pathological and Hemodynamic Features in Dementia Subtypes
| Feature | Alzheimer’s Disease (AD) | Vascular Dementia (VaD) | Mixed Dementia |
| Primary proteinopathy | Amyloid-β plaques; tau tangles | Minimal or absent | Extensive Aβ and tau |
| Vascular lesions | Often present (CAA, microbleeds) | Large infarcts, lacunae, WMH | Combined vascular and proteinopathic |
| Hemodynamics | Reduced CBF; glymphatic failure | Focal or global ischemia | Chronic hypoperfusion + stiffness |
| Lipid driver | ApoB link via cerebrovascular injury | Strong LDL-C / ApoB association | Lifelong high ApoB burden |
| Imaging hallmarks | Hippocampal atrophy; Aβ-PET (+) | Multiple infarcts; carotid stenosis | Atrophy, WMH, plaque burden |
Adapted from references [3,5,9,32,40]. CAA = cerebral amyloid angiopathy; WMH = white-matter hyperintensities; CBF = cerebral blood flow.
Lipoprotein Dynamics and Lifelong Causal Exposure
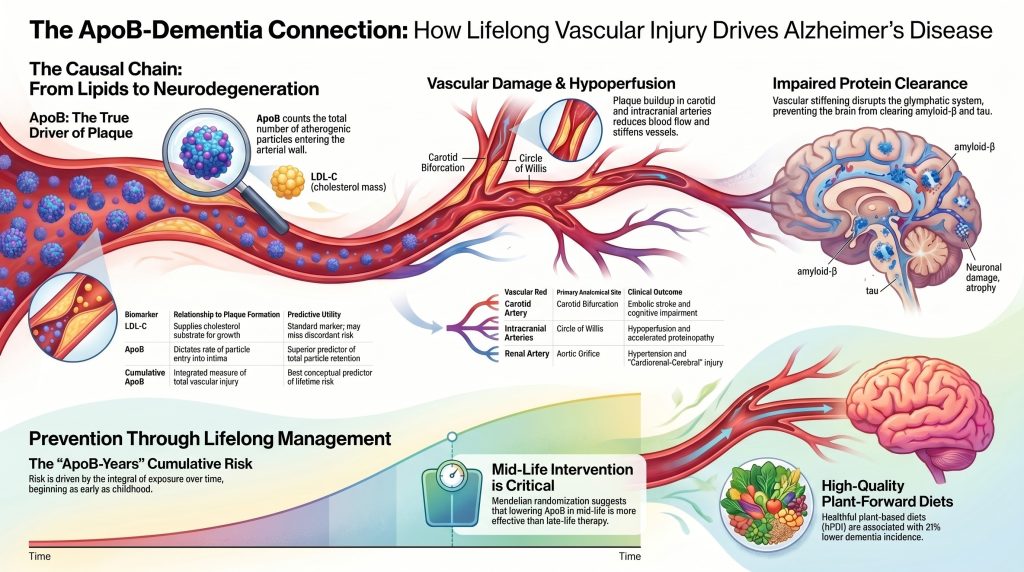
Central to the development of atherosclerosis is the concentration of ApoB-containing lipoproteins in the circulation.[10,19,20] ApoB is a structural protein found on the surface of every atherogenic lipoprotein particle. Hepatically derived very-low-density lipoproteins (VLDL), intermediate-density lipoproteins (IDL), low-density lipoproteins (LDL), and lipoprotein(a) each carry one molecule of apoB-100 per particle, while chylomicrons and chylomicron remnants carry apoB-48 originating from the intestine.[20] In adults in the post-absorptive state, the dominant atherogenic burden reflected by plasma ApoB measurement is overwhelmingly composed of apoB-100–bearing particles.[20] Beyond the count distinction, LDL particles (apoB-100) have substantially longer intimal residence times than larger, more transiently circulating chylomicron remnants (apoB-48), so the apoB-100 fraction contributes disproportionately to cumulative arterial-wall injury and dominates the “ApoB-years” integral in the cerebrovascular bed.
Whereas LDL cholesterol (LDL-C) measures the cholesterol mass within LDL particles, ApoB provides a direct count of total atherogenic particles.[20] This distinction is mechanistically critical: the probability of a lipoprotein particle entering the arterial intima and being retained by extracellular proteoglycans is a function of particle concentration rather than cholesterol content per particle.[19] Multivariable Mendelian randomization analyses have established that ApoB is the dominant lipid trait in the relationship between lipoproteins and coronary heart disease; when ApoB is conditioned upon, neither LDL-C nor triglycerides retain independent causal information for atherosclerotic cardiovascular disease.[21,25]
The risk conferred by ApoB is cumulative — a concept now formalized as the LDL/ApoB cumulative exposure hypothesis.[22] Atherogenesis is a slow process that typically begins in childhood with the formation of nascent fatty streaks.[19] As ApoB-containing particles remain elevated over decades, atherosclerotic plaque burden gradually accrues until critical thresholds are crossed, leading to luminal narrowing or plaque rupture.[22,33] Lifelong cumulative exposure (often described as “cholesterol-years” or “ApoB-years”) is a more biologically appropriate predictor of cardiovascular and, plausibly, dementia risk than any single mid-life or late-life measurement, which may be confounded by reverse causation in older adults developing frailty and weight loss in the preclinical phase of dementia.[22,24,28]
Table 2. Comparative Predictive Value of Lipid Biomarkers for Atherosclerotic Risk
| Biomarker | Definition | Relationship to plaque formation | Utility |
| LDL-C | Cholesterol mass in LDL particles | Supplies cholesterol substrate for plaque growth | Standard marker; misses risk in discordance |
| ApoB | Total count of atherogenic particles (one per particle) | Dictates rate of particle entry into the intima | Superior predictor of particle retention |
| Non-HDL-C | Total cholesterol minus HDL-C | Captures LDL, VLDL, IDL, and Lp(a) cholesterol | Practical surrogate for atherogenic burden |
| Lp(a) | LDL-like particle with apolipoprotein(a) | Pro-thrombotic and pro-inflammatory | Independent genetic risk factor |
| Cumulative ApoB exposure | Integral of ApoB over time (“ApoB-years”) | Integrated measure of total vascular injury | Best conceptual predictor of lifetime risk |
Adapted from references [19,20,21,22,25,33].
Review of Mendelian Randomization Evidence in Dementia
Mendelian randomization (MR) leverages the random assortment of genetic alleles during gametogenesis to estimate the causal effect of modifiable exposures on outcomes, mimicking the design of a randomized controlled trial while avoiding many sources of observational confounding.[29] All MR claims summarized below have been evaluated against the STROBE-MR statement on transparent reporting of Mendelian randomization investigations.[29]
MR analyses of genetic instruments for common lipid-lowering drug targets — HMGCR (statin target), PCSK9 (PCSK9 inhibitor target), NPC1L1 (ezetimibe target), and CETP — provide some of the strongest current causal evidence linking lifelong lower non-HDL-C to lower dementia risk. In a one-sample MR meta-analysis of 1,091,775 individuals, the odds ratios per 1 mmol/L (≈39 mg/dL) lower non-HDL-C for all-cause dementia were 0.24 (95% CI 0.18–0.31) for HMGCR, 0.18 (0.12–0.25) for NPC1L1, 0.30 (0.26–0.34) for CETP, and 0.97 (0.70–1.35) for PCSK9.[7] An earlier Danish MR analysis reported risk ratios per 1 mmol/L lower LDL-C of 0.57 (0.27–1.17) for Alzheimer’s disease and 0.81 (0.34–1.89) for vascular dementia — directionally consistent with protection but underpowered for individual outcomes.[27]
Two points warrant explicit acknowledgement. First, the primary outcome in Nordestgaard et al. is all-cause dementia rather than biomarker-confirmed Alzheimer’s disease under the 2024 NIA-AA criteria.[40] Second, the PCSK9 estimate is compatible with the null, in contrast to robust signals for HMGCR, NPC1L1, and CETP.[7] This target-specific heterogeneity argues against a simple model in which any pharmacologic mechanism that lowers ApoB-containing lipoproteins will equivalently lower dementia risk, and points toward target-specific cerebrovascular biology that future trials must address.
MR evidence specifically for ApoB and Alzheimer’s disease is more recent. Univariable two-sample MR using UK Biobank ApoB instruments and IGAP AD summary statistics shows higher genetically proxied ApoB is associated with shortened healthspan and — in the authors’ own hedged language — may possibly increase risk for Alzheimer’s disease.[4] Multivariable MR conditioning on LDL-C indicates that ApoB retains its association with shortened healthspan even when LDL-C is null in the joint model.[4] We adopt the authors’ own characterization: this is supportive but preliminary causal evidence at the AD endpoint, which should be replicated against biomarker-confirmed AD outcomes as such data become available.
Limitations of Mendelian Randomization
Several limitations apply to all MR evidence presented above and must inform causal interpretation:
- Survival bias: individuals with genetically high ApoB may die of premature cardiovascular events before reaching the age at which dementia would clinically manifest, potentially attenuating the observed lipid–AD association.[4,7]
- Competitive risk of death: more specifically than general survival bias, ApoB is so strongly causal for myocardial infarction and stroke that individuals with genetically elevated ApoB who would otherwise have developed AD are disproportionately removed from the AD-eligible population by earlier cardiovascular mortality. This competing-risk structure systematically attenuates the observable lipid–AD association in standard Mendelian randomization designs, and is one specific reason the AD-endpoint signal in current MR is weaker than the cardiovascular signal — not necessarily because the causal pathway is weak.[4,27]
- Pleiotropy: genetic variants may affect the outcome through pathways other than the intended exposure. MR-Egger, weighted median, and multivariable MR estimators were used by the studies cited to assess robustness; ApoB-conditional analyses retain a directionally consistent signal.[4,21,25]
- Outcome heterogeneity: clinically diagnosed AD in administrative datasets frequently includes substantial vascular copathology;[3,32] biomarker-confirmed AD endpoints under the 2024 NIA-AA framework[40] would strengthen future MR work. The drug-target MR evidence for all-cause dementia[7] is methodologically stronger but applies to dementia broadly, not to biomarker-confirmed AD specifically.
- Ancestry and generalizability: most MR studies of lipids and dementia have been conducted in populations of European ancestry; causal estimates may differ across ancestry groups.[7]
- Lifelong-exposure interpretation: MR estimates the cumulative effect of lifelong genetic exposure, which typically exceeds the effect achievable by pharmacotherapy initiated in late life. This difference is informative for prevention timing but does not directly translate to expected effect sizes from late-life dementia trials.[22,26]
Regional Pathophysiology of Atherosclerotic Plaque Formation
Atherosclerosis is a chronic inflammatory disease initiated by lipid retention in focal areas of arteries, particularly regions of disturbed, non-laminar flow.[10,19] The molecular and cellular mechanisms of plaque formation are broadly conserved across major vascular beds, yet local anatomy and hemodynamics impart distinctive characteristics to disease in each location.[10,11,31]
Carotid Artery Atherosclerosis
The carotid bifurcation is especially vulnerable to plaque formation owing to its complex geometry and flow reversal during the cardiac cycle.[10,11] Low and oscillatory wall shear stress at the origin of the internal carotid artery promotes endothelial dysfunction and increases vessel-wall permeability to ApoB-containing lipoproteins.[10,11,19] A distinctive feature of carotid plaque is its tendency to embolize: high-velocity flow dislodges thrombi forming on ulcerated plaque surfaces, generating recurrent microemboli rather than complete proximal occlusion.[11,34] Cumulative microembolic injury, lacunar infarction, and microbleeds are core substrates of vascular cognitive impairment.[2,9,34]
Intracranial Atherosclerotic Disease (ICAD)
ICAD refers to plaque formation in the large arteries at the base of the brain — including the middle cerebral artery, basilar artery, and the Circle of Willis.[11,13] Unlike extracranial carotid disease, ICAD has been directly linked, in prospective longitudinal imaging, to subsequent deposition of amyloid-β and tau in the brain parenchyma. In a prospective cohort with serial PiB-PET (amyloid) and AV-1451-PET (tau) imaging, the presence of any intracranial stenosis at baseline was significantly associated with greater amyloid-β accumulation over four years, and stenosis in two or more arteries predicted greater tau deposition over two years.[13] This longitudinal directionality — vascular injury preceding accelerated proteinopathy — is among the strongest current human imaging evidence that intracranial vascular health is causally upstream of AD-defining pathology rather than merely co-occurring with it.
Renal Artery Atherosclerosis and the Cardiorenal-Cerebral Axis
Renal artery stenosis (RAS) is overwhelmingly atherosclerotic in origin and concentrates at the aortic orifice or within the proximal one-third of the main renal artery.[31,37] The kidney, like the brain, is a low-resistance vascular bed sensitive to hyperpulsatile flow and ischemia.[6,12] Plaque progression in the renal artery is augmented by dysfunctional perivascular adipose tissue and expansion of the vasa vasorum, which release pro-inflammatory cytokines and extracellular vesicles.[31]
Atherosclerotic RAS activates the renin-angiotensin-aldosterone system, producing secondary hypertension that further accelerates damage to the cerebral microvasculature and establishes a feed-forward loop linking kidney, heart, and brain.[6,31,36,39] Chronic kidney disease is itself characterized by systemic inflammation, oxidative stress, and uremic endothelial toxicity, all of which are independently associated with cognitive decline through the cardiorenal-cerebral axis.[6,38,39]
Table 3. Comparison of Plaque Characteristics Across Vascular Beds
| Vascular bed | Primary anatomical site | Unique hemodynamic driver | Primary clinical outcome |
| Carotid artery | Carotid bifurcation; ICA origin | Flow reversal and turbulence; low/oscillatory shear | Embolic stroke and cognitive impairment |
| Intracranial arteries | MCA, basilar, Circle of Willis | Complex branching; loss of autoregulation | Hypoperfusion and accelerated proteinopathy |
| Renal artery | Aortic orifice / proximal one-third | Orifice-related shear stress | RAS, hypertension, CKD |
| Coronary artery | Proximal LAD and bifurcations | Cyclic compressive stress; bending | Myocardial infarction and angina |
Adapted from references [10,11,13,31,37].
The Mechanistic Pathway: From Lipids to Neurodegeneration
The transition from elevated circulating ApoB to clinical dementia involves a multi-stage cascade in which subendothelial retention of ApoB-containing lipoproteins serves as the initiating event.[19] Retained particles undergo oxidative and enzymatic modification, generating potent pro-inflammatory signals that activate endothelial cells, recruit monocytes, and drive macrophage foam-cell formation.[10,19,53]
Lipid Retention, Endothelial Activation, and Hypoperfusion
Endothelial cells respond to oxidized lipoproteins by expressing adhesion molecules (VCAM-1, ICAM-1) and releasing chemokines (MCP-1, CXCL1) and cytokines (IL-1β, IL-6).[10,19,53] Progressive plaque growth produces arterial narrowing and stiffening,[10,12] which in the cerebrovascular tree manifests as reduced cerebral blood flow and chronic ischemia.[12,17] Chronic hypoperfusion induces metabolic stress in neurons and glia, activates the NLRP3 inflammasome, and primes the brain for accelerated neurodegeneration.[17,53]
Amyloid-β in the Vascular Pathway
A central element of the vascular framing is that amyloid-β (Aβ) accumulation is, in part, both a cause and a consequence of cerebrovascular injury.[16,17] Ischemia and oxidative stress upregulate amyloid precursor protein expression and shift its processing toward the amyloidogenic pathway,[16,17] while vascular Aβ deposition (cerebral amyloid angiopathy, CAA) impairs vessel-wall contractility and reduces protein efflux from the brain.[16] This generates a feed-forward loop: vascular injury promotes Aβ accumulation, which causes additional vascular damage and neuroinflammation through innate immune activation.[16,17,53]
We are explicit, however, that amyloid is not merely downstream debris from vascular injury. Autosomal-dominant familial AD caused by mutations in APP, PSEN1, or PSEN2 establishes amyloid as a sufficient pathogenic factor in its own right — these mutations produce AD through altered amyloid biology without requiring upstream vascular insult.[41] Anti-amyloid monoclonal antibody trials in sporadic AD, discussed below in the Counterarguments section, further demonstrate that amyloid removal produces statistically robust clinical benefit, which is incompatible with a model in which amyloid is epiphenomenal. Our claim is therefore narrower and more defensible: in sporadic late-onset AD — the overwhelming majority of cases — decades of ApoB-driven cerebrovascular injury are a major upstream determinant of the cerebral environment in which intrinsic amyloid and tau biology become pathogenic. The vascular and amyloid pathways are interacting and bidirectionally amplifying rather than mutually exclusive.
Glymphatic Dysfunction and Protein Clearance
Clearance of metabolic waste from the brain is mediated in part by the glymphatic system, a perivascular CSF–interstitial fluid exchange network whose driving force depends on arterial pulsatility and stable venous outflow.[15,18] Arterial stiffening — a direct mechanical consequence of atherosclerosis and vascular aging — impairs the pulsatile driving force,[12] and rodent and human work have shown that hypertension and aging reduce CSF influx along the perivascular space.[18] When glymphatic clearance is impaired, neurotoxic proteins including Aβ and tau accumulate, contributing to synaptic dysfunction and cognitive decline.[12,15,18] We treat the arterial-stiffness → impaired-glymphatic-clearance → AD chain as a mechanistically plausible and partially supported hypothesis rather than an established clinical pathway. Human glymphatic physiology remains incompletely validated, and the diffusion-tensor-image analysis along the perivascular space (DTI-ALPS) index is an imaging proxy for perivascular diffusivity rather than a direct measure of glymphatic flux; its reproducibility and biological specificity are the subject of active methodological work.[35]
Testing the Lifelong Protection Hypothesis
Early, sustained lipid lowering is critical to preventing irreversible neurodegeneration.[22,26] MR studies effectively provide a natural model of lifelong lower LDL-C and ApoB, showing that individuals with genetically lower exposure carry substantially lower lifetime cardiovascular risk than those who initiate lipid-lowering therapy only in late life.[22,25]
Quantitatively, clinical trials in the Cholesterol Treatment Trialists’ Collaboration demonstrate approximately a 22% relative reduction in major vascular events per 1 mmol/L (≈39 mg/dL) reduction in LDL-C achieved with statin therapy, irrespective of baseline risk.[26] By contrast, MR estimates a markedly larger reduction in coronary heart disease per 1 mmol/L genetically lower lifelong LDL-C — roughly threefold the per-mmol/L benefit observed with late-life statin therapy.[22,33] We label this difference clearly as an inference about the value of duration: it argues that for dementia prevention, interventions should ideally occur in midlife, before mature plaques and the cerebrovascular substrate for cognitive decline have developed.[22,40] Direct extrapolation of the cardiovascular per-mmol/L estimate to dementia outcomes is not warranted in the absence of trial data, and we present the timing inference rather than a numerical prediction of dementia-event reduction.
Randomized vascular-risk-reduction trials provide complementary, if smaller-effect, evidence. In SPRINT MIND, intensive blood-pressure control (target SBP <120 mmHg) versus standard control (<140 mmHg) reduced the composite of mild cognitive impairment or probable dementia (hazard ratio 0.85, 95% CI 0.74–0.97) and reduced MCI specifically (HR 0.81, 95% CI 0.69–0.94), although the probable-dementia primary endpoint did not reach statistical significance.[14] In the FINGER multidomain prevention trial, a two-year combined intervention of diet, exercise, cognitive training, and vascular risk monitoring improved cognitive composite scores versus control in at-risk older adults.[50] These trials demonstrate that randomized modification of vascular risk factors can produce measurable cognitive benefit, supporting the broader logic of lifelong vascular-risk reduction for dementia prevention.
The Role of Plant-Based Dietary Patterns
High-quality plant-based dietary patterns are a powerful non-pharmacological strategy for reducing lifelong ApoB exposure and systemic vascular inflammation.[23,46,49] The quality of the diet matters: a healthful plant-based diet index (hPDI) emphasizing whole grains, fruits, vegetables, legumes, and nuts is associated with lower dementia risk, whereas an unhealthful plant-based pattern (uPDI) high in refined grains and sugar-sweetened beverages is associated with higher risk.[43,46]
Meta-analytic evidence from prospective cohorts (n≈208,000) shows the highest tertile of hPDI is associated with approximately 21% lower dementia incidence relative to the lowest tertile, while the highest tertile of uPDI is associated with approximately 26% higher dementia incidence.[43] In adults with established cardiometabolic disease — a population for whom the cardiorenal-cerebral axis is already activated — a healthful plant-based diet pattern combined with broader healthy-lifestyle factors substantially attenuates dementia risk in UK Biobank follow-up.[45] Among plant-forward dietary patterns, Mediterranean and MIND diets currently have the strongest cumulative evidence base, including randomized cardiovascular outcome data (PREDIMED)[49] and consistent observational associations with lower dementia risk;[44] strict whole-food plant-based or vegan patterns have weaker direct evidence for dementia or AD endpoints specifically. We therefore frame plant-forward dietary patterns broadly as a biologically well-grounded prevention strategy supported by strong observational data and (for Mediterranean diet) randomized cardiovascular evidence, and we do not assert the superiority of strict whole-food plant-based or vegan diets over Mediterranean or MIND patterns on the basis of currently available dementia data.
Beyond ApoB lowering itself, neuroprotective effects of high-quality plant-based dietary patterns are mediated by complementary mechanisms: improved endothelial function and reduced oxidative stress;[23,46] favorable modulation of the gut microbiome with reduced production of trimethylamine N-oxide (TMAO), a metabolite linked to atherosclerosis and cognitive impairment;[47,48] and improved blood pressure, insulin sensitivity, and inflammatory tone.[23,49,50] Mediterranean and MIND dietary patterns share many of these mechanisms and are supported by both observational and randomized evidence for cardiovascular outcomes and cognitive endpoints.[44,49,50]
Table 4. Impact of Dietary Patterns on Vascular and Cognitive Risk Factors
| Dietary pattern | Key components | Effect on ApoB | Effect on inflammation | Dementia risk |
| Healthful plant-based (hPDI) | Whole plants, legumes, nuts | Decreased | Decreased | Lower [43,45] |
| Mediterranean | Olive oil, plants, fish, nuts | Moderately decreased | Moderately decreased | Lower [44,49] |
| MIND | Plants, berries, fish, olive oil | Moderately decreased | Decreased | Lower [44] |
| Unhealthful plant-based (uPDI) | Refined grains, sweets, fried potatoes | Variable / neutral | Increased | Higher [43] |
| Western | Red and processed meat, refined sugars | Increased | Increased | Higher [23,46] |
Adapted from references [23,43,44,45,46,49].
Counterarguments and Reconciliation with the Biological Definition of AD
The hypothesis that ApoB-driven atherosclerosis is a causal upstream contributor to Alzheimer’s disease must engage three legitimate counterarguments: (1) the late-life low-cholesterol paradox, (2) cases of neurodegeneration without overt vascular disease, and (3) the partial therapeutic success of anti-amyloid monoclonal antibodies, which appear to support an amyloid-causal model.
The Late-Life Low-Cholesterol Paradox
Observational studies frequently show that low cholesterol in adults aged ≥ 75 years is associated with higher dementia risk.[24,28] The most parsimonious explanation is reverse causation: preclinical dementia is preceded by years of unintentional weight loss and frailty, and in late-stage neurodegeneration sometimes by hepatic synthetic dysfunction, all of which lower circulating cholesterol independent of any protective effect of low lifelong ApoB exposure.[24,28] This paradox underscores why midlife cholesterol measurements and lifelong genetic instruments — not geriatric cholesterol levels — are the appropriate substrate for causal inference about the ApoB–dementia relationship.[22,29]
Non-Vascular AD and Genetic Architecture
Predominantly neurodegenerative Alzheimer’s disease occurs through specific autosomal-dominant mutations in APP, PSEN1, and PSEN2,[41] and additional contributors include chronic infection, mitochondrial dysfunction, sleep-disordered breathing, and chronic sleep deprivation.[41] These are not refutations of a vascular-causal model; they are simply reminders that AD is a heterogeneous syndrome in which atherosclerosis is a major causal contributor in a substantial subset of patients, not the exclusive cause in all.[3,32,41] The APOE ε4 allele increases AD risk in significant part through cerebrovascular mechanisms — including blood-brain barrier breakdown that precedes cognitive decline,[42] impaired Aβ clearance, and increased CAA[16] — illustrating that genetic and vascular risk are intertwined rather than competing.
Anti-Amyloid Monoclonal Antibody Trials
Lecanemab and donanemab provide the most direct test of the amyloid pathway in symptomatic disease. In CLARITY AD, lecanemab slowed CDR-SB decline by 0.45 points relative to placebo over 18 months on an 18-point scale (mean 1.21 vs 1.66; 27% relative slowing), with amyloid-related imaging abnormalities (ARIA-E) in 12.6% and symptomatic ARIA-E in 2.8%.[51] In TRAILBLAZER-ALZ 2, donanemab slowed iADRS decline by 35% and CDR-SB decline by 36% over 76 weeks; ARIA-E occurred in 24.0% (symptomatic 6.1%), with three donanemab-related deaths attributable to ARIA.[52]
We accept these trials as substantive evidence that amyloid is mechanistically important in symptomatic AD: removing amyloid produces measurable, statistically robust, and consistent slowing of cognitive decline across primary and secondary endpoints. That result is not compatible with a model in which amyloid is epiphenomenal vascular debris, and we do not adopt such a model. Whether the absolute magnitude of slowing reaches a minimal clinically important difference (MCID) is genuinely contested rather than settled, with proposed CDR-SB MCIDs for early AD ranging across roughly 0.5–2.0 points and different working groups landing in different places along that range. Reasonable people disagree on whether the lecanemab and donanemab effects clear that threshold.
What the trials do not show is that amyloid removal in late-stage symptomatic disease is sufficient to halt or reverse AD progression: the effects are incremental, the ARIA safety profile is non-trivial and concentrated in APOE ε4 homozygotes, and the trials test late-stage amyloid removal rather than upstream prevention. These results are fully compatible with our framing — amyloid is mechanistically important and a legitimate therapeutic target; decades of upstream ApoB-driven cerebrovascular injury are an additional and modifiable major determinant of the substrate on which amyloid pathology operates; and a complete dementia-prevention strategy reasonably invests in both pathways.
Engagement with the 2024 NIA-AA Biological Definition of AD
The 2024 Alzheimer’s Association revised criteria operationalize AD biologically by core amyloid and tau biomarkers, independent of clinical syndrome.[40] These criteria are a diagnostic framework, not an etiologic theory. The working group does not claim that amyloid and tau are the sole upstream causes of AD and does not deny vascular contributions — indeed, vascular and other co-pathologies are explicitly recognized as common modifiers of disease course. We are therefore not arguing against the 2024 criteria; we are making a complementary etiologic argument compatible with them. For a substantial subset of patients meeting the biological criteria for AD, the upstream cause of the amyloid/tau biomarker fingerprint is, in our reading, lifelong ApoB-driven cerebrovascular atherogenesis interacting with intrinsic amyloid biology. Longitudinal evidence that intracranial atherosclerosis predicts subsequent amyloid and tau accumulation[13] is directly compatible with this reading.
Proposed Research Designs to Test the Hypothesis
Rigorous testing of the vascular-atherosclerotic causal hypothesis requires longitudinal studies that integrate cardiovascular and neurodegenerative markers across the life course.[9,40] Studies should follow individuals from their 30s and 40s with serial measurement of ApoB, Lp(a), and inflammatory markers.[20,22] High-resolution imaging — including contrast-enhanced ultrasound and vessel-wall MRI — should monitor plaque growth and regression in the carotid and intracranial arteries.[11,13] Advanced neuroimaging, including amyloid- and tau-PET and emerging perivascular-diffusivity proxies of glymphatic function, would map how vascular injury precedes protein accumulation.[13,15,18,35]
New MR studies should isolate ApoB from other lipid traits using multivariable instruments and examine its independent effect against biomarker-confirmed AD endpoints under the 2024 NIA-AA criteria.[4,40] Randomized prevention trials are needed to test whether early, aggressive ApoB lowering — initiated in midlife rather than in late life — reduces subsequent cognitive decline and neurodegenerative-protein burden.[7,22,26] Trials should stratify by APOE ε4 status and baseline cerebrovascular burden,[16,42] and should comply with STROBE-MR or CONSORT reporting as appropriate.[29]
Final Thesis and Conclusion
Cognitive decline, vascular cognitive impairment and dementia, mixed-pathology dementia, and — in a substantial subset of patients — biologically defined Alzheimer’s disease can be productively understood as late neurological manifestations of decades-long vascular-lipid-inflammatory injury operating alongside intrinsic amyloid and tau biology.[1,2,3,17,40] The central modifiable driver of this vascular pathway is lifelong exposure to ApoB-containing lipoproteins, which initiates and propagates atherosclerotic plaque formation in critical cerebral-supplying and renal arteries.[7,20,22,31] These plaques cause chronic cerebral hypoperfusion, arterial stiffening, blood-brain barrier dysfunction, and impaired glymphatic clearance, contributing to the accumulation of amyloid and tau and to clinical cognitive decline in interaction with intrinsic neurodegenerative biology.[12,13,16,17,18,42]
Our argument is therefore not that Alzheimer’s disease should be reclassified as vascular disease or that amyloid is mechanistically unimportant. Familial AD,[41] anti-amyloid trial results in sporadic AD,[51,52] and the 2024 NIA-AA biological criteria[40] each establish that amyloid biology is a legitimate and necessary component of any complete model of AD. Our argument is more focused: for a substantial subset of patients meeting biological criteria for AD, the upstream cause of the amyloid/tau fingerprint is, in important part, decades of ApoB-driven cerebrovascular injury. This framing is fully compatible with the 2024 NIA-AA biological criteria, which define AD diagnostically by amyloid and tau biomarkers without making etiologic claims about the upstream cause of those biomarkers.
The practical consequence is that prevention strategy for Alzheimer’s disease and related dementias should expand to give substantially greater weight to lifelong preservation of vascular integrity — through high-quality plant-forward dietary patterns,[43,44,45] randomized vascular-risk-reduction strategies such as those validated in SPRINT MIND and FINGER,[14,50] and, where indicated, lipid-lowering pharmacotherapy initiated in midlife rather than in late life[7,22,26] — alongside continued investment in late-stage protein-targeted therapy. Both arms of intervention are warranted by current evidence; the upstream vascular arm offers the larger plausible public-health gain on the longest time horizons, and is the focus of this manuscript.
References
- Iadecola C. The pathobiology of vascular dementia. Neuron. 2013;80(4):844-866. doi:10.1016/j.neuron.2013.10.008
- Smith EE, Aparicio HJ, Gottesman RF, et al. Vascular Contributions to Cognitive Impairment and Dementia in the United States: Prevalence and Incidence: A Scientific Statement From the American Heart Association. Stroke. 2025;56(10):e317-e330. doi:10.1161/STR.0000000000000494
- Kapasi A, DeCarli C, Schneider JA. Impact of multiple pathologies on the threshold for clinically overt dementia. Acta Neuropathol. 2017;134(2):171-186. doi:10.1007/s00401-017-1717-7
- Martin L, Boutwell BB, Messerlian C, Adams CD. Mendelian randomization reveals apolipoprotein B shortens healthspan and possibly increases risk for Alzheimer’s disease. Commun Biol. 2024;7(1):230. Published 2024 Feb 24. doi:10.1038/s42003-024-05887-2
- Sweeney MD, Sagare AP, Zlokovic BV. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol. 2018;14(3):133-150. doi:10.1038/nrneurol.2017.188
- Bugnicourt JM, Godefroy O, Chillon JM, Choukroun G, Massy ZA. Cognitive disorders and dementia in CKD: the neglected kidney-brain axis. J Am Soc Nephrol. 2013;24(3):353-363. doi:10.1681/ASN.2012050536
- Nordestgaard LT, Hanson A, Sanderson E, et al. Cholesterol-lowering drug targets reduce risk of dementia: Mendelian randomization and meta-analyses of 1 million individuals. Alzheimers Dement. 2025;21(10):e70638. doi:10.1002/alz.70638
- Bos D, Vernooij MW, de Bruijn RF, et al. Atherosclerotic calcification is related to a higher risk of dementia and cognitive decline. Alzheimers Dement. 2015;11(6):639-47.e1. doi:10.1016/j.jalz.2014.05.1758
- Wardlaw JM, Smith C, Dichgans M. Small vessel disease: mechanisms and clinical implications. Lancet Neurol. 2019;18(7):684-696. doi:10.1016/S1474-4422(19)30079-1
- Libby P, Buring JE, Badimon L, et al. Atherosclerosis. Nat Rev Dis Primers. 2019;5(1):56. Published 2019 Aug 16. doi:10.1038/s41572-019-0106-z
- Yahagi K, Kolodgie FD, Lutter C, et al. Pathology of Human Coronary and Carotid Artery Atherosclerosis and Vascular Calcification in Diabetes Mellitus. Arterioscler Thromb Vasc Biol. 2017;37(2):191-204. doi:10.1161/ATVBAHA.116.306256
- Mitchell GF, van Buchem MA, Sigurdsson S, et al. Arterial stiffness, pressure and flow pulsatility and brain structure and function: the Age, Gene/Environment Susceptibility–Reykjavik study. Brain. 2011;134(Pt 11):3398-3407. doi:10.1093/brain/awr253
- Kang KM, Park C, Nam H, et al. Intracranial stenosis and longitudinal progression of Alzheimer’s disease pathologies. Alzheimers Dement. 2025;21(12):e71010. doi:10.1002/alz.71010
- SPRINT MIND Investigators for the SPRINT Research Group, Williamson JD, Pajewski NM, et al. Effect of Intensive vs Standard Blood Pressure Control on Probable Dementia: A Randomized Clinical Trial. JAMA. 2019;321(6):553-561. doi:10.1001/jama.2018.21442
- Iliff JJ, Wang M, Liao Y, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci Transl Med. 2012;4(147):147ra111. doi:10.1126/scitranslmed.3003748
- Greenberg SM, Bacskai BJ, Hernandez-Guillamon M, Pruzin J, Sperling R, van Veluw SJ. Cerebral amyloid angiopathy and Alzheimer disease – one peptide, two pathways. Nat Rev Neurol. 2020;16(1):30-42. doi:10.1038/s41582-019-0281-2
- Iadecola C. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron. 2017;96(1):17-42. doi:10.1016/j.neuron.2017.07.030
- Mestre H, Tithof J, Du T, et al. Flow of cerebrospinal fluid is driven by arterial pulsations and is reduced in hypertension. Nat Commun. 2018;9(1):4878. Published 2018 Nov 19. doi:10.1038/s41467-018-07318-3
- Tabas I, Williams KJ, Borén J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 2007;116(16):1832-1844. doi:10.1161/CIRCULATIONAHA.106.676890
- Soffer DE, Marston NA, Maki KC, et al. Role of apolipoprotein B in the clinical management of cardiovascular risk in adults: An Expert Clinical Consensus from the National Lipid Association. J Clin Lipidol. 2024;18(5):e647-e663. doi:10.1016/j.jacl.2024.08.013
- Richardson TG, Sanderson E, Palmer TM, et al. Evaluating the relationship between circulating lipoprotein lipids and apolipoproteins with risk of coronary heart disease: A multivariable Mendelian randomisation analysis. PLoS Med. 2020;17(3):e1003062. Published 2020 Mar 23. doi:10.1371/journal.pmed.1003062
- Ference BA, Braunwald E, Catapano AL. The LDL cumulative exposure hypothesis: evidence and practical applications. Nat Rev Cardiol. 2024;21(10):701-716. doi:10.1038/s41569-024-01039-5
- Sacks FM, Lichtenstein AH, Wu JHY, et al. Dietary Fats and Cardiovascular Disease: A Presidential Advisory From the American Heart Association. Circulation. 2017;136(3):e1-e23. doi:10.1161/CIR.0000000000000510
- Mielke MM, Zandi PP, Sjögren M, et al. High total cholesterol levels in late life associated with a reduced risk of dementia. Neurology. 2005;64(10):1689-1695. doi:10.1212/01.WNL.0000161870.78572.A5
- Zuber V, Gill D, Ala-Korpela M, et al. High-throughput multivariable Mendelian randomization analysis prioritizes apolipoprotein B as key lipid risk factor for coronary artery disease. Int J Epidemiol. 2021;50(3):893-901. doi:10.1093/ije/dyaa216
- Cholesterol Treatment Trialists’ (CTT) Collaboration, Baigent C, Blackwell L, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376(9753):1670-1681. doi:10.1016/S0140-6736(10)61350-5
- Benn M, Nordestgaard BG, Frikke-Schmidt R, Tybjærg-Hansen A. Low LDL cholesterol, PCSK9 and HMGCR genetic variation, and risk of Alzheimer’s disease and Parkinson’s disease: Mendelian randomisation study. BMJ. 2017;357:j1648. Published 2017 Apr 24. doi:10.1136/bmj.j1648
- Iwagami M, Qizilbash N, Gregson J, et al. Blood cholesterol and risk of dementia in more than 1·8 million people over two decades: a retrospective cohort study. Lancet Healthy Longev. 2021;2(8):e498-e506. doi:10.1016/S2666-7568(21)00150-1
- Skrivankova VW, Richmond RC, Woolf BAR, et al. Strengthening the Reporting of Observational Studies in Epidemiology Using Mendelian Randomization: The STROBE-MR Statement. JAMA. 2021;326(16):1614-1621. doi:10.1001/jama.2021.18236
- Schwartz GG, Steg PG, Szarek M, et al. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N Engl J Med. 2018;379(22):2097-2107. doi:10.1056/NEJMoa1801174
- Safian RD. Renal artery stenosis. Prog Cardiovasc Dis. 2021;65:60-70. doi:10.1016/j.pcad.2021.03.003
- Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology. 2007;69(24):2197-2204. doi:10.1212/01.wnl.0000271090.28148.24
- Domanski MJ, Tian X, Wu CO, et al. Time Course of LDL Cholesterol Exposure and Cardiovascular Disease Event Risk. J Am Coll Cardiol. 2020;76(13):1507-1516. doi:10.1016/j.jacc.2020.07.059
- Bos D, Arshi B, van den Bouwhuijsen QJA, et al. Atherosclerotic Carotid Plaque Composition and Incident Stroke and Coronary Events. J Am Coll Cardiol. 2021;77(11):1426-1435. doi:10.1016/j.jacc.2021.01.038
- Taoka T, Ito R, Nakamichi R, et al. Reproducibility of diffusion tensor image analysis along the perivascular space (DTI-ALPS) for evaluating interstitial fluid diffusivity and glymphatic function: CHanges in Alps index on Multiple conditiON acquIsition eXperiment (CHAMONIX) study. Jpn J Radiol. 2022;40(2):147-158. doi:10.1007/s11604-021-01187-5
- Toyoda K. Cerebral small vessel disease and chronic kidney disease. J Stroke. 2015;17(1):31-37. doi:10.5853/jos.2015.17.1.31
- Hansen KJ, Edwards MS, Craven TE, et al. Prevalence of renovascular disease in the elderly: a population-based study. J Vasc Surg. 2002;36(3):443-451. doi:10.1067/mva.2002.127351
- Sedaghat S, Sorond F, Yaffe K, et al. Decline in kidney function over the course of adulthood and cognitive function in midlife. Neurology. 2020;95(17):e2389-e2397. doi:10.1212/WNL.0000000000010631
- Drew DA, Weiner DE, Sarnak MJ. Cognitive Impairment in CKD: Pathophysiology, Management, and Prevention. Am J Kidney Dis. 2019;74(6):782-790. doi:10.1053/j.ajkd.2019.05.017
- Jack CR Jr, Andrews JS, Beach TG, et al. Revised criteria for diagnosis and staging of Alzheimer’s disease: Alzheimer’s Association Workgroup. Alzheimers Dement. 2024;20(8):5143-5169. doi:10.1002/alz.13859
- Knopman DS, Amieva H, Petersen RC, et al. Alzheimer disease. Nat Rev Dis Primers. 2021;7(1):33. Published 2021 May 13. doi:10.1038/s41572-021-00269-y
- Montagne A, Nation DA, Sagare AP, et al. APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature. 2020;581(7806):71-76. doi:10.1038/s41586-020-2247-3
- Shen J, Chen H, Gong Y, et al. Association between plant-based diets and incident dementia: results from prospective cohort studies and a meta-analysis. J Prev Alzheimers Dis. 2026;13(2):100457. doi:10.1016/j.tjpad.2025.100457
- de Crom TOE, Mooldijk SS, Ikram MK, Ikram MA, Voortman T. MIND diet and the risk of dementia: a population-based study. Alzheimers Res Ther. 2022;14(1):8. Published 2022 Jan 12. doi:10.1186/s13195-022-00957-1
- Dunk MM, Dove A, Wang J, Sakakibara S, Carballo-Casla A, Xu W. Plant-Based Diet Quality, Healthy Lifestyle, and Dementia Risk in Older Adults With Cardiometabolic Diseases. JACC Adv. 2025;4(11 Pt 1):102229. doi:10.1016/j.jacadv.2025.102229
- Satija A, Bhupathiraju SN, Spiegelman D, et al. Healthful and Unhealthful Plant-Based Diets and the Risk of Coronary Heart Disease in U.S. Adults. J Am Coll Cardiol. 2017;70(4):411-422. doi:10.1016/j.jacc.2017.05.047
- Tang WH, Wang Z, Levison BS, et al. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med. 2013;368(17):1575-1584. doi:10.1056/NEJMoa1109400
- Vogt NM, Romano KA, Darst BF, et al. The gut microbiota-derived metabolite trimethylamine N-oxide is elevated in Alzheimer’s disease. Alzheimers Res Ther. 2018;10(1):124. Published 2018 Dec 22. doi:10.1186/s13195-018-0451-2
- Estruch R, Ros E, Salas-Salvadó J, et al. Primary Prevention of Cardiovascular Disease with a Mediterranean Diet Supplemented with Extra-Virgin Olive Oil or Nuts. N Engl J Med. 2018;378(25):e34. doi:10.1056/NEJMoa1800389
- Ngandu T, Lehtisalo J, Solomon A, et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. Lancet. 2015;385(9984):2255-2263. doi:10.1016/S0140-6736(15)60461-5
- van Dyck CH, Swanson CJ, Aisen P, et al. Lecanemab in Early Alzheimer’s Disease. N Engl J Med. 2023;388(1):9-21. doi:10.1056/NEJMoa2212948
- Sims JR, Zimmer JA, Evans CD, et al. Donanemab in Early Symptomatic Alzheimer Disease: The TRAILBLAZER-ALZ 2 Randomized Clinical Trial. JAMA. 2023;330(6):512-527. doi:10.1001/jama.2023.13239
- Heneka MT, Carson MJ, El Khoury J, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14(4):388-405. doi:10.1016/S1474-4422(15)70016-5