
Cardiovascular Mechanisms of Longitudinal Performance Decline in Master Athletes:
A Narrative Review of Aging, Coronary Artery Disease, Silent Myocardial Ischemia, and Unrecognized Myocardial Infarction
Abstract
This narrative review synthesizes selected peer-reviewed evidence on the cardiovascular and physiological mechanisms underlying longitudinal performance decline in master athletes between the ages of 50 and 80. Topics addressed include (1) the natural trajectory of physical capacity from the Swedish Population Cohort for Physical Activity and Fitness (SPAF) study[1]; (2) the contrast between master-athlete and sedentary V̇O2max decline[2][3][4]; (3) central and peripheral cardiovascular mechanisms of V̇O2max erosion; (4) the paradox of subclinical coronary atherosclerosis in lifelong endurance athletes; (5) silent myocardial ischemia and unrecognized myocardial infarction — including epidemiology, mechanisms, prognosis, and diagnostic approach; (6) coronary microvascular dysfunction as a candidate contributor to exercise intolerance in selected older athletes; and (7) contemporary sports-cardiology screening considerations grounded in the 2020 ESC Sports Cardiology Guideline. The synthesis is interpretive rather than exhaustive; specific search strategy and scope are described in the Literature Identification section.
1. Introduction
The aging process in humans is inexorably linked to a progressive decline in physiological capacity, yet the rate and trajectory of this decline are significantly influenced by habitual physical activity. Among individuals who maintain high levels of endurance training into their sixth, seventh, and eighth decades — commonly referred to as master athletes — the decline in maximal aerobic capacity (V̇O2max) and endurance performance provides a unique biological model for studying the limits of human aging[2]. While master athletes consistently demonstrate superior cardiorespiratory fitness (CRF) compared to their sedentary peers, they are not immune to the fundamental biological processes that erode cardiovascular function over time[3][4].
Longitudinal performance decline between the ages of 50 and 80 is governed by a complex interplay of central cardiac limitations, peripheral vascular and mitochondrial decay, and the paradoxical emergence of subclinical cardiovascular pathologies. These include coronary artery disease in lifelong endurance athletes[13][14][15] and, in the broader aging population, silent myocardial ischemia (SMI), unrecognized myocardial infarction (UMI), and coronary microvascular dysfunction (CMD)[21][22]. To understand these mechanisms in context, it is helpful first to establish a population-level baseline from the SPAF longitudinal cohort[1] and then to contrast that baseline with the specific physiological adaptations — and pathological risks — observed in lifelong endurance athletes.
2. Literature Identification
This article is a narrative review. Literature was identified primarily through PubMed and Embase searches (1990–2026) and by backward reference review of relevant primary studies, meta-analyses, and major-society guidance documents addressing master athletes, age-related decline in aerobic capacity, coronary atherosclerosis in endurance athletes, silent myocardial ischemia, unrecognized myocardial infarction, coronary microvascular dysfunction, and sports-cardiology screening. Priority was given to original cohort studies, randomized trials, systematic reviews, meta-analyses, and official guideline statements. Because this was not a protocol-driven systematic review, study selection was qualitative and the synthesis should be interpreted as interpretive rather than exhaustive. No formal meta-analytic pooling was performed; quantitative values are reproduced from the cited primary sources without re-analysis.
3. The General Population Baseline: Insights from the Westerståhl SPAF Study
The 47-year longitudinal SPAF study reported by Westerståhl and colleagues offers one of the most comprehensive assessments of how physical capacity changes from adolescence into early old age in a representative general-population cohort[1]. Tracking 427 individuals born in 1958, the study used repeated objective assessments of aerobic capacity, muscular endurance, and explosive power from age 16 to age 63[1]. The dataset is informative because it identifies the natural rate of decline in a population where physical activity is not standardized, highlighting the accelerating nature of biological aging regardless of elite athletic status.
Peak physical performance was generally attained between the ages of 26 and 36 for both absolute aerobic capacity and muscular endurance[1]. Specifically, men reached their peak absolute aerobic capacity at age 35 (3.26 L·min−1), while women peaked at age 36 (2.61 L·min−1)[1]. After these peaks, decline began at approximately 0.3–0.6% per year and accelerated to roughly 2.0–2.5% per year as participants approached their 60s[1]. By age 63, cumulative decline in absolute aerobic capacity from peak was approximately 33% in men and 30% in women[1].
Table 1. Physical capacity trajectories from the 47-year SPAF longitudinal cohort.
| Parameter | Men (peak age 35–36) | Women (peak age 36) | Total decline by 63 |
| Absolute aerobic capacity (L·min⁻¹) | 3.26 | 2.61 | 30–33% |
| Relative aerobic capacity (mL·kg⁻¹·min⁻¹) | 42.2 at age 26 | 39.7 at age 31 | 37–40% |
| Vertical jump (cm) | 45.5 at age 27 | 33.0 at age 19 | 41–48% |
| Muscular endurance (bench press, reps) | 52.7 at age 36 | 39.7 at age 34 | 32–35% |
Source: Westerståhl M, et al. J Cachexia Sarcopenia Muscle. 2025[1]. Values are cohort means at the indicated age. SPAF = Swedish Population Cohort for Physical Activity and Fitness.
The SPAF data also underscored the role of lifestyle factors in modifying these trajectories. Higher leisure-time physical activity at age 16 and an active lifestyle during adulthood were associated with superior performance across all metrics throughout life[1]. Higher educational attainment was associated with higher absolute aerobic capacity and muscular endurance, plausibly via health-seeking behaviors. Even with these protective factors, however, the acceleration of decline after age 40 was a consistent finding, reinforcing the concept that biological aging imposes a progressive physiological constraint on functional capacity that begins to operate well before it becomes clinically significant.
4. Comparative Decline: Master Athletes vs. General Population
When contrasting the SPAF baseline with longitudinal data from master athletes, a clear distinction emerges in starting altitude and slope of V̇O2max decline. Master endurance athletes represent a cohort that has maximized adaptive potential and may carry a V̇O2max at age 60 that exceeds the median for a sedentary 40-year-old[2]. Despite this advantage, the inevitable decline in V̇O2max remains the primary physiological mechanism associated with reduced endurance performance in aging athletes[3][4].
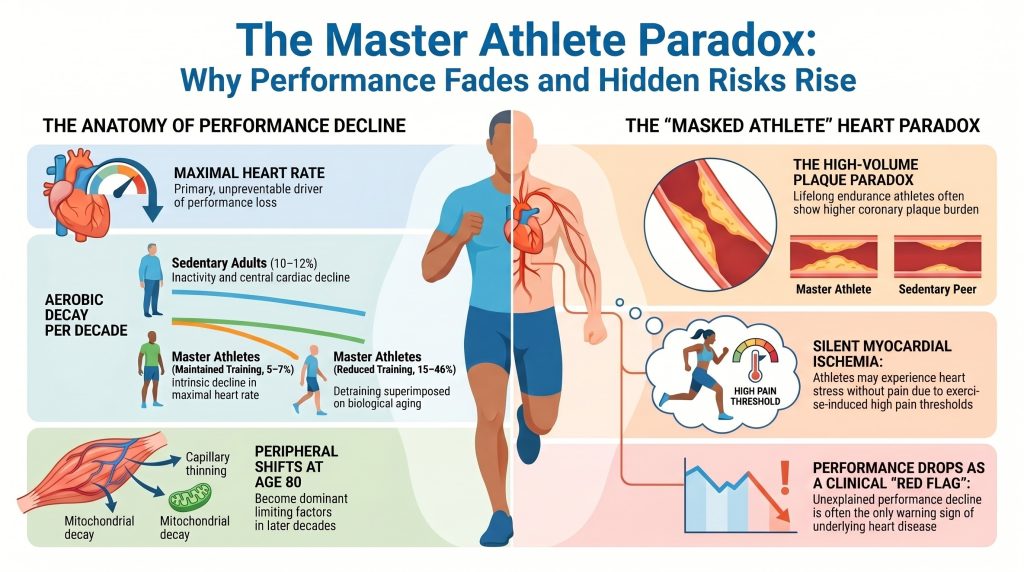
Longitudinal assessments indicate that master athletes who maintain high-intensity, high-volume training reduce the rate of V̇O2max decline to approximately 5–7% per decade, compared with 10–12% per decade in age-matched sedentary individuals[2][3][4]. This training-attenuated decline is, however, fragile; regression analyses indicate that a substantial proportion of the variance in V̇O2max decline in master athletes is directly attributable to reductions in training volume and intensity, with steepest declines (15–46% per decade) observed in athletes who substantially reduce training[3][4].
Table 2. Decadal rates of V̇O2max decline by group.
| Group | Decadal decline | Primary driver |
| Sedentary adults | 10–12% | Sarcopenia, inactivity, central cardiac decline |
| Master athletes (maintained training) | 5–7% | Intrinsic decline in maximal heart rate |
| Master athletes (reduced training) | 15–46% | Detraining superimposed on aging |
Sources: refs [2][3][4].
5. Cardiovascular Mechanisms of Aerobic Capacity Erosion
The erosion of V̇O2max is mathematically described by the Fick principle: V̇O2max = Q̇ × (a–v)O2 difference, where Q̇ is cardiac output and (a–v)O2 is the arteriovenous oxygen content difference. In master athletes, the relative contribution of central cardiac factors (heart rate and stroke volume) versus peripheral factors (oxygen extraction) shifts with age[8][9].
5.1 Central mechanisms: the aging pump
The most consistent and largely unmodifiable driver of V̇O2max decline is the age-related reduction in maximal heart rate (HRmax). The widely cited Tanaka equation — HRmax ≈ 208 − 0.7 × age — implies an average decline of approximately 0.7 beats·min−1 per year[7]. This is driven primarily by changes in the intrinsic electrophysiology of the sinoatrial node and by reduced β-adrenergic responsiveness with age. While endurance training can lower resting heart rate and enhance parasympathetic tone, it has limited ability to preserve maximal heart rate during all-out exertion.
Stroke volume (SV) provides a more nuanced story. In sedentary individuals, SV declines due to increased myocardial stiffness, reduced ventricular compliance, and impaired diastolic filling[10]. In master athletes, chronic endurance training induces eccentric ventricular remodeling characterized by larger chamber dimensions and enhanced myocardial compliance[10][11]. This adaptation allows athletes to maintain higher SV through the Frank-Starling mechanism. Nonetheless, master athletes can experience reduced SV at peak exercise due to abnormal shortening of diastolic filling time at very high heart rates, where the diastolic interval is too short for complete ventricular filling.
5.2 Peripheral mechanisms: oxygen extraction and mitochondrial decay
While central limitations dominate the early phases of decline, peripheral factors become increasingly limiting as athletes approach age 80[9]. Three peripheral processes are most consequential:
- Arteriovenous O2 Maximal (a–v)O2 difference declines modestly with age in master athletes, largely driven by capillary rarefaction — thinning of the capillary networks supplying individual muscle fibers — which increases diffusion distance for oxygen[9].
- Mitochondrial function. Aging is associated with reductions in mitochondrial density and in the activity of key oxidative enzymes such as citrate synthase and succinate dehydrogenase[2]. Maintenance of high-intensity training in master athletes preserves mitochondrial quality, but reductions in training volume produce a rapid, near-linear drop in oxidative capacity.
- Selective atrophy of Type II (fast-twitch) fibers and overall muscle-mass loss reduces metabolic demand and exercise output, an effect documented in the SPAF cohort and elsewhere[1].
6. Coronary Atherosclerosis in Master Athletes: The Calcification Paradox
One of the most challenging findings in modern sports cardiology is the apparent paradox of high coronary artery calcium (CAC) scores in lifelong endurance athletes. Cross-sectional data, including the Master@Heart cohort, have shown that lifelong endurance athletes (predominantly male) often carry a higher prevalence of coronary plaques and higher CAC scores than sedentary peers of similar risk profile[13][14][15][16][17].
6.1 Plaque morphology and distribution
The Master@Heart study by De Bosscher and colleagues enrolled 191 lifelong athletes, 191 late-onset athletes, and 176 healthy non-athletes (all male) and demonstrated that lifelong athletes had the highest coronary plaque burden[15]. The burden included not only calcified plaques — generally considered stable — but also non-calcified and mixed plaques in proximal coronary segments[15].
Table 3. Coronary plaque findings from the Master@Heart cohort.
| Group | ≥1 coronary plaque (OR) | ≥1 proximal plaque (OR) | ≥50% luminal stenosis |
| Non-athletes (reference) | 1.00 | 1.00 | ≈ reference |
| Late-onset athletes | intermediate | intermediate | low |
| Lifelong endurance athletes | 1.86 | 1.96 | uncommon, but elevated vs. controls |
Source: De Bosscher R, et al. Eur Heart J. 2023[15]. OR = odds ratio versus non-athlete reference; values are adjusted estimates. All participants were male; mean age ≈ 60 years. Obstructive (≥50%) stenosis was uncommon in absolute terms across all groups; exact reported prevalence in the lifelong-athlete arm should be cross-checked against the primary publication before final typesetting.
In Master@Heart, lifelong athletes were also disproportionately likely to have non-calcified plaques in proximal segments compared with non-athletes[15]. Proximal location is clinically relevant because such plaques control flow to large myocardial territories. While higher cardiorespiratory fitness is associated with stable plaque morphology in some analyses, the absolute presence of these lesions suggests that high-volume endurance training may interact with conventional risk factors to accelerate progression of pre-existing subclinical disease[16][17].
6.2 Proposed mechanistic drivers
The development of CAD in master athletes remains incompletely explained. The current observational literature raises several proposed mechanisms, which should be interpreted as plausible hypotheses rather than established causal pathways:
- Hemodynamic shear stress. Vigorous exercise produces hyperdynamic coronary flow. At sites of disturbed or turbulent flow, mechanical forces may contribute to endothelial dysfunction[13][17].
- Oxidative stress. Extreme metabolic demands of ultra-endurance training may transiently generate reactive oxygen species in excess of antioxidant capacity, with possible downstream effects on endothelial function and lipoprotein retention[17].
- Exercise-induced hypertension. In a recent cohort of male master endurance athletes, occult resting and exercise-induced hypertension was prevalent and associated with higher coronary plaque burden[18]. Exaggerated exercise systolic blood pressure responses are increasingly recognized as a clinically actionable phenotype.
- Interactions with conventional risk factors. Even in athletes, conventional risk factors — LDL-cholesterol, apolipoprotein B, lipoprotein(a), hypertension, family history — remain the dominant drivers of atherosclerosis[49][50].
7. Silent Myocardial Ischemia and Unrecognized Myocardial Infarction
7.1 Definitions and key distinction
Silent myocardial ischemia (SMI) refers to objective evidence of transient inducible ischemia — typically ST-segment depression on ambulatory or exercise ECG, or inducible perfusion abnormality on stress imaging — in the complete absence of anginal symptoms[21][22]. Unrecognized myocardial infarction (UMI) refers to a completed infarction that was not clinically diagnosed at the time it occurred[28][29][30][31]. Late gadolinium enhancement (LGE) on cardiac magnetic resonance (CMR) characterizes prior myocardial injury — scar, fibrosis, or completed infarction — and is therefore the reference standard imaging modality for UMI rather than a primary test for active ischemia[30][36]. This distinction is preserved throughout the remainder of this section.
7.2 Epidemiology of silent ischemia and unrecognized MI
Silent or unrecognized MIs account for a substantial fraction of all myocardial infarctions in epidemiological cohorts. The original Framingham Heart Study reported that approximately one-quarter of MIs were unrecognized when first detected during routine biennial examination[28]. The Atherosclerosis Risk in Communities (ARIC) study found that the proportion of MIs that were silent and detected only by ECG was approximately 22%, with substantial sex and race differences in detection[29]. In the Cardiovascular Health Study of older community-dwelling adults, the prevalence of UMI rose with age[31].
The advent of cardiac MRI with LGE has substantially raised estimates of UMI prevalence. The ICELAND-MI study by Schelbert and colleagues found that UMI detected by CMR-LGE was more than twice as common as UMI detected by ECG in an older community cohort, and that ECG-detected UMI represented only a fraction of all CMR-detected events[30]. In the MESA cohort, CMR-detected myocardial scar was identified in approximately 7.9% of asymptomatic adults aged 45–84 with no clinical history of MI[33].
Translated to US epidemiology, of the approximately 805,000 myocardial infarctions occurring annually in the United States, ECG-based community surveillance suggests that roughly 170,000 (≈21%) are clinically silent or unrecognized at the time of occurrence — a figure consistent with ARIC and Framingham proportions[28][29][31]. Because CMR-based detection has identified more than twice as many UMIs as ECG in older community cohorts[30], the ECG-based extrapolation of 170,000 is best interpreted as a lower bound on the true annual burden of clinically unrecognized myocardial infarction in the United States.
7.3 Why some myocardial ischemia and infarction is silent: proposed mechanisms
The absence of pain during ischemia or infarction is multifactorial. Eight broad mechanisms have been proposed in the literature:
- Autonomic neuropathy. Afferent sympathetic and vagal fibers that transmit cardiac nociceptive signals can be damaged by diabetes mellitus, age-related autonomic neuropathy, or prior cardiac surgery, attenuating or eliminating the perception of ischemic pain[21].
- Higher pain threshold. Individuals vary substantially in baseline somatic pain thresholds, and a subset of patients with documented ischemia have demonstrably elevated thresholds for laboratory pain stimuli[34].
- Endogenous opioid and endocannabinoid systems. Circulating β-endorphin and related opioids are elevated in some patients with silent ischemia and have been implicated in central modulation of cardiac pain perception[34].
- Exercise-induced hypoalgesia (EIH). Regular endurance training engages the endogenous opioid and endocannabinoid systems and is consistently associated with elevated pain thresholds and tolerances in laboratory testing[25][26]. In master athletes, EIH plausibly contributes to the absence of warning symptoms during demand-induced ischemia.
- Small infarct size and limited transmural extent. Small subendocardial or non-transmural infarcts may not generate sufficient afferent signaling to cross the threshold for conscious perception[32].
- Anatomic distribution. Inferior-wall ischemia and posterior-wall ischemia are more frequently silent than anterior-wall events, possibly due to differences in afferent innervation density[21].
- Collateral circulation. Chronic exercise promotes the development of coronary collateral vessels, which can maintain blood flow distal to a stenosis at rest and during low-to-moderate exertion, thereby preventing ischemia and pain until peak demand exceeds collateral capacity[27].
- Ischemic preconditioning. Repeated brief episodes of subclinical ischemia may protect the myocardium against subsequent insults and may also blunt afferent pain signaling[35].
7.4 Symptoms — and the absence of symptoms
By definition, classic exertional angina is absent in silent ischemia and unrecognized MI. However, the term silent is sometimes a misnomer. On careful retrospective questioning, a proportion of patients with apparently silent events describe subtle symptoms at the time — most commonly unaccustomed dyspnea, exertional fatigue out of proportion to workload, atypical chest discomfort attributed to indigestion or musculoskeletal causes, syncope or near-syncope, or unexplained reduction in exercise tolerance[21][22][32]. These so-called anginal equivalents are particularly important in master athletes, in whom dropping performance — rather than chest pain — may be the only clinical warning. When no symptoms are reported in any form, the event is genuinely asymptomatic and is detected only through screening, incidental imaging, or as part of a subsequent workup.
7.5 Prognosis
Silent ischemia and unrecognized MI are not benign. In long-term follow-up of the ICELAND-MI cohort, UMI detected by CMR was associated with mortality comparable to that of clinically recognized MI[30]. Across multiple cohorts, individuals with UMI carry elevated risks of heart failure, recurrent MI, and sudden cardiac death[29][30][31]. In endurance athletes specifically, the presence of ischemic-pattern LGE has been associated with markedly elevated risk of subsequent major adverse cardiac events including sudden cardiac death[36].
7.6 Diagnosis
Diagnostic approaches differ for SMI versus UMI:
- For silent ischemia, the foundational modalities are ambulatory (Holter) ECG, exercise ECG, and stress imaging (stress echocardiography, single-photon emission CT, positron emission tomography, or stress CMR perfusion)[21][22].
- For unrecognized MI, the resting 12-lead ECG has limited sensitivity, detecting only a fraction of CMR-confirmed events[30]. CMR with LGE is the most sensitive non-invasive test and is the reference standard for non-acute scar detection[30][36]. Echocardiographic regional wall-motion abnormalities and elevated high-sensitivity troponin in non-acute contexts may also raise suspicion.
7.7 Master-athlete-specific burden: the “masked athlete” phenomenon
In a seminal cross-sectional study, Katzel and colleagues found that approximately 16% of male master athletes (mean age 60 years) had silent ischemia on a symptom-limited graded exercise test, a prevalence statistically comparable to that observed in sedentary controls of similar age[23]. Athletic status did not reduce the prevalence of ischemic burden; rather, it changed the perception of that burden. Combined with the EIH literature[25][26], these findings support the concept of the masked athlete — an individual whose high cardiorespiratory fitness and elevated pain thresholds can defer the perception of ischemia until exertional demand approaches the limits of coronary supply.
CMR-LGE prevalence in master endurance athletes has been variably reported, ranging from approximately 11% in younger marathon runners to considerably higher figures in older endurance-athlete cohorts[37][38]. Ischemic-pattern LGE (subendocardial enhancement in a coronary distribution) and non-ischemic LGE (focal patchy enhancement at the right-ventricular hinge points or mid-myocardium) carry different mechanistic implications and different prognostic weights[36]. A sudden or unexplained decline in performance or in V̇O2max in a previously stable master athlete should therefore be treated as a clinical red flag warranting consideration of advanced imaging.
8. Coronary Microvascular Dysfunction
Coronary microvascular dysfunction (CMD) is a plausible contributor to exercise intolerance in some older athletes, but the relevant evidence base in master-athlete cohorts is limited and findings should be described cautiously[39][40]. In a selected cohort of athletes with abnormal exercise tests but no obstructive epicardial CAD on coronary CT angiography, Foulkes and colleagues reported significantly lower coronary flow reserve (3.3 vs 4.2 in controls) and elevated endothelin-1 levels[39]. Several mechanisms have been proposed in athletes specifically, including the hypothesis of a possible “capillary-to-myocyte mismatch” in which training-induced hypertrophy outpaces microvascular expansion. These findings are best interpreted as preliminary and hypothesis-generating rather than as evidence that CMD broadly explains performance decline across the master-athlete population.
CMD may present with atypical features, including reduced exercise capacity, unusually elevated heart rates during submaximal effort, and exertional dyspnea rather than classic chest pain[40]. Diagnosis typically requires specialized testing (PET-based coronary flow reserve, invasive coronary physiology with bolus thermodilution, or stress CMR) available at experienced centers.
9. Screening and Risk Stratification in the Master Athlete
Given the high prevalence of subclinical CAD and silent ischemia in older athletes, traditional screening tools have important limitations. The resting 12-lead ECG, while valuable for detecting electrical disorders, has low sensitivity for subclinical atherosclerosis[45]. Exercise treadmill testing in asymptomatic athletes is constrained by high false-positive rates[44]. Importantly, individual tests should be selected according to the clinical question being asked — anatomy, inducible ischemia, prior scar, or microvascular function — rather than by pooled performance metrics across these distinct domains.
Table 4. Cardiac diagnostic modalities in older master athletes, organized by the clinical question each test answers.
| Modality | Primary question answered | Main strength | Main limitation |
| Resting 12-lead ECG | Electrical disease; prior Q-wave MI | Widely available baseline test | Low sensitivity for silent CAD or non-Q-wave prior MI |
| Exercise treadmill ECG | Exercise-provoked ECG abnormalities; symptom correlation | Functional provocation; reveals exercise BP response | Both false positives and false negatives are common in master athletes: the latter because high cardiorespiratory reserve and collateral flow may allow some athletes to maintain workload despite inducible ischemia until higher exercise intensities |
| Coronary CT angiography (CCTA) | Coronary anatomy; plaque burden and stenosis | Strong anatomic definition of plaque/stenosis | Does not directly establish inducible ischemia |
| Stress echocardiography / stress perfusion imaging (SPECT, PET, stress CMR) | Inducible ischemia | Functional assessment of ischemic burden | Performance depends on protocol and image quality |
| CMR with LGE | Prior infarction / scar / fibrosis pattern (UMI) | High tissue characterization value; gold standard for non-acute scar | Not a primary test for obstructive CAD |
| PET with coronary flow reserve | Inducible ischemia plus microvascular function | Useful for CMD assessment in specialized centers | Limited availability; higher complexity |
Sources: refs [40][44][45]. CCTA = coronary CT angiography; CMR = cardiac magnetic resonance; LGE = late gadolinium enhancement; UMI = unrecognized myocardial infarction; CMD = coronary microvascular dysfunction; SPECT = single-photon emission computed tomography; PET = positron emission tomography. The table is intentionally structured by clinical question rather than by pooled sensitivity/specificity, because tests answering different reference-standard questions are not directly comparable.
The ISCHEMIA trial should not be summarized as proving that exercise capacity outweighs coronary anatomy or ischemia severity. A more defensible conclusion from the ISCHEMIA program is that, in stable chronic coronary disease, an initial invasive strategy did not produce an overall reduction in major cardiovascular events compared with an initial conservative strategy, and prognosis varied according to disease burden in pre-specified analyses[41][42]. The broader point that cardiorespiratory fitness is a strong predictor of mortality is robustly supported by cardiopulmonary exercise testing literature outside the ISCHEMIA program[43]. For master athletes, the practical implication is that a sudden or unexplained decline in performance or in V̇O2max should be treated as a clinical signal warranting structured evaluation[47].
10. Sex Differences and the Female Master Athlete
A substantial portion of the master-athlete coronary literature is derived from male cohorts, including Master@Heart, which enrolled only men[15]. In female master athletes, CAC prevalence is generally lower than in male counterparts at the same age and training exposure, and the coronary phenotype associated with lifelong endurance training is less well characterized[48]. The 2020 ESC Sports Cardiology Guideline emphasizes individualized risk assessment in female master athletes, with attention to traditional risk factors, menopausal status, and exercise blood pressure response[47]. Generalizing male-cohort findings to female master athletes is inappropriate until parallel data are available.
11. Clinical Principles for the Aging Master Athlete
Current evidence supports individualized training and cardiovascular risk management rather than a single proven training formula for preventing coronary plaque in master athletes. Practical principles supported by primary literature and major-society guidance include:
- Maintain regular endurance activity at intensities and volumes appropriate to the individual, recognizing that detraining accelerates V̇O2max decline beyond what biological aging alone produces[2][3].
- Incorporate resistance training to help preserve muscle mass and function across the aging trajectory.
- Aggressively manage conventional cardiovascular risk factors — LDL-cholesterol or apolipoprotein B, lipoprotein(a) where available, blood pressure, glycemia, and tobacco exposure — in accordance with current prevention guidelines[45][49][50].
- Evaluate exaggerated exercise blood pressure responses, which are independently associated with higher coronary plaque burden in master athletes[18].
- Treat unexplained performance decline as a clinical signal warranting structured assessment, given the “masked athlete” phenomenon[23][25].
- Apply advanced imaging (CCTA, stress imaging, CMR with LGE) selectively, guided by clinical context, exercise findings, and conventional risk profile[47].
Polarized training distributions (commonly described as approximately 80/20 low- to high-intensity) may be useful performance optimization frameworks but have not been prospectively shown to prevent CAC accrual or to mitigate the so-called athlete paradox, and should not be presented as preventive prescriptions[51][52].
12. Synthesis and Discussion
12.1 Inevitable versus modifiable decay
The SPAF study confirms that the rise and fall of physical capacity is a constitutive feature of human life[1]. The age of peak performance and the acceleration of decline after age 40 appear largely invariant across populations. However, master athletes demonstrate that the absolute level of performance and the decadal rate of V̇O2max loss are highly modifiable through maintenance of training volume and intensity[2][3][4].
12.2 Dominance of central factors
Between ages 50 and 80, the primary driver of performance decline in master athletes is central: a relentless decline in HRmax reduces total cardiac output during peak exertion, a limitation that even aggressive training cannot reverse[7]. Although athletes preserve stroke volume better than sedentary peers, age-related myocardial stiffening eventually restricts diastolic filling at high heart rates[10][11].
12.3 The pathological paradox
Lifelong endurance training appears associated with both favorable cardiovascular adaptation and an increased prevalence of subclinical coronary atherosclerosis in some observational cohorts[15][16][17]. The atherosclerotic burden — combined with the analgesic effects of exercise-induced hypoalgesia — makes silent ischemia and unrecognized MI clinically important risks that warrant individualized screening and management[23][25][26].
12.4 Peripheral mechanisms at the limit of human aging
As athletes approach age 80, peripheral extraction increasingly catches up to central factors as a determinant of V̇O2max. Capillary rarefaction and mitochondrial decay reduce the muscle’s capacity to utilize delivered oxygen — an effect that is accelerated when training volume is reduced[2][9].
Key Clinical Takeaways
| • Aerobic capacity declines along a curvilinear trajectory beginning in the mid-30s and accelerating in the 60s. Master athletes who maintain training reduce the rate of decline to roughly half that of sedentary peers; detraining accelerates decline several-fold.
• Lifelong endurance training does not confer immunity to coronary atherosclerosis. Master@Heart and related cohorts show higher coronary calcium and proximal plaque burden in lifelong athletes than in sedentary peers; plaque morphology may be more favorable but does not abolish risk. • Silent ischemia and unrecognized myocardial infarction are not uncommon in older athletes. Approximately 1 in 6 male master athletes has inducible ischemia on graded exercise testing; CMR-detected scar in community cohorts exceeds ECG-detected scar by more than 2:1. • Symptoms may be absent, atypical, or expressed as performance loss. An unexplained decline in race times, exertional dyspnea, or reduced exercise tolerance in an older athlete should prompt structured cardiac evaluation — not reassurance. • Choose the diagnostic test to match the clinical question. Anatomy (CCTA), inducible ischemia (stress imaging), prior scar (CMR with LGE), and microvascular function (PET with CFR) answer different questions and are not interchangeable. • Standard cardiovascular risk-factor management remains the foundation. Aggressively manage LDL-C/ApoB, lipoprotein(a) where available, blood pressure (including exaggerated exercise responses), glycemia, and tobacco exposure. No training distribution has been prospectively shown to prevent CAC accrual. • Overall health benefits of lifelong exercise remain substantial. The clinical task is calibration of intensity, surveillance, and risk-factor control — not avoidance of training. |
13. Conclusions
For master athletes between the ages of 50 and 80, maintenance of endurance performance requires a careful balance between sustained training (to preserve V̇O2max and skeletal-muscle oxidative capacity) and individualized cardiovascular risk management. Transitioning from active to sedentary status produces a V̇O2max decline substantially steeper than aging alone[2][3].
However, the masked athlete concept warrants vigilance rather than complacency. Older endurance athletes may present with reduced performance capacity, exertional dyspnea, or other atypical symptoms rather than classic angina, and unexplained decline in performance should prompt structured clinical reassessment[23][25][47].
The available literature supports substantial overall health benefits of long-term exercise[43][53]. Some evidence suggests that the plaque phenotype of lifelong athletes is shifted toward more calcified and fewer mixed/non-calcified lesions when matched for total plaque burden, which may confer relative stability compared with sedentary patients of equivalent CAC score[16]. Even granting that observation, however, the present evidence base does not justify claiming that coronary calcification in master athletes is uniformly benign, or that mortality advantages persist regardless of plaque phenotype — CAC-stratified mortality data specific to master athletes remain limited. A more evidence-consistent conclusion is that lifelong exercise and subclinical coronary disease can coexist, that plaque morphology likely matters as well as plaque quantity, and that individualized risk assessment is essential in older athletes.
14. Limitations
This is a narrative review. No systematic search protocol or formal meta-analytic pooling was performed. The Master@Heart cohort and several other key coronary-imaging studies in lifelong endurance athletes are all-male, limiting generalizability to female master athletes. CAC-stratified mortality data specific to master athletes remain sparse, and the prognostic significance of non-calcified proximal plaques identified incidentally on CCTA in asymptomatic athletes is not yet established. The relevant CMD literature in master athletes is derived from small selected cohorts on referral and should not be generalized.
Disclosures and End-Matter
Conflicts of interest
The author reports no financial conflicts of interest related to the subject matter of this manuscript. The author is the operator of CuringHeartDisease.com, a clinician-educator platform that provides peer-reviewed cardiovascular health content without supplement marketing or commercial sponsorship.
Funding
No external funding, sponsorship, or institutional support contributed to the conception, drafting, or publication of this manuscript.
Data availability
No new datasets were generated for this narrative review. All cited evidence is contained in the published literature and is accessible through the cited references.
Ethics
Ethics approval and informed consent were not required because this manuscript is a review of previously published literature and does not report new human-subject research.
Prior dissemination
No version of this manuscript has been previously published or made publicly available in any venue, including preprint servers and the author’s educational platform. CuringHeartDisease.com is the intended publication venue.
Author contributions
PM conceived the manuscript, conducted the literature identification, drafted and revised the text, and approved the final version for publication.
Acknowledgments
The author thanks the external reviewers whose comments informed the present revision.
References
References are listed in order of first mention in the text, in accordance with NLM/Vancouver convention. All entries are PubMed-indexed primary studies, meta-analyses, systematic reviews, or major-society guidelines. Author lists with seven or more authors are truncated after the first six followed by “et al.”
- Westerståhl M, Jörnåker G, Jansson E, et al. Rise and Fall of Physical Capacity in a General Population: A 47-Year Longitudinal Study. J Cachexia Sarcopenia Muscle. 2025;16(6):e70134. doi:10.1002/jcsm.70134
- Trappe S. Master athletes. Int J Sport Nutr Exerc Metab. 2001;11 Suppl:S196-S207. doi:10.1123/ijsnem.11.s1.s196
- Pollock ML, Mengelkoch LJ, Graves JE, et al. Twenty-year follow-up of aerobic power and body composition of older track athletes. J Appl Physiol (1985). 1997;82(5):1508-1516. doi:10.1152/jappl.1997.82.5.1508
- Tanaka H, Seals DR. Endurance exercise performance in Masters athletes: age-associated changes and underlying physiological mechanisms. J Physiol. 2008;586(1):55-63. doi:10.1113/jphysiol.2007.141879
- Hawkins S, Wiswell R. Rate and mechanism of maximal oxygen consumption decline with aging: implications for exercise training. Sports Med. 2003;33(12):877-888. doi:10.2165/00007256-200333120-00002
- Joyner MJ, Coyle EF. Endurance exercise performance: the physiology of champions. J Physiol. 2008;586(1):35-44. doi:10.1113/jphysiol.2007.143834
- Tanaka H, Monahan KD, Seals DR. Age-predicted maximal heart rate revisited. J Am Coll Cardiol. 2001;37(1):153-156. doi:10.1016/s0735-1097(00)01054-8
- Levine BD. .VO2max: what do we know, and what do we still need to know?. J Physiol. 2008;586(1):25-34. doi:10.1113/jphysiol.2007.147629
- Proctor DN, Joyner MJ. Skeletal muscle mass and the reduction of VO2max in trained older subjects. J Appl Physiol (1985). 1997;82(5):1411-1415. doi:10.1152/jappl.1997.82.5.1411
- Pelliccia A, Caselli S, Sharma S, et al. European Association of Preventive Cardiology (EAPC) and European Association of Cardiovascular Imaging (EACVI) joint position statement: recommendations for the indication and interpretation of cardiovascular imaging in the evaluation of the athlete’s heart. Eur Heart J. 2018;39(21):1949-1969. doi:10.1093/eurheartj/ehx532
- La Gerche A, Burns AT, Mooney DJ, et al. Exercise-induced right ventricular dysfunction and structural remodelling in endurance athletes. Eur Heart J. 2012;33(8):998-1006. doi:10.1093/eurheartj/ehr397
- Levine BD. Can intensive exercise harm the heart? The benefits of competitive endurance training for cardiovascular structure and function. Circulation. 2014;130(12):987-991. doi:10.1161/CIRCULATIONAHA.114.008142
- Aengevaeren VL, Mosterd A, Braber TL, et al. Relationship Between Lifelong Exercise Volume and Coronary Atherosclerosis in Athletes. Circulation. 2017;136(2):138-148. doi:10.1161/CIRCULATIONAHA.117.027834
- Möhlenkamp S, Lehmann N, Breuckmann F, et al. Running: the risk of coronary events : Prevalence and prognostic relevance of coronary atherosclerosis in marathon runners. Eur Heart J. 2008;29(15):1903-1910. doi:10.1093/eurheartj/ehn163
- De Bosscher R, Dausin C, Claus P, et al. Lifelong endurance exercise and its relation with coronary atherosclerosis. Eur Heart J. 2023;44(26):2388-2399. doi:10.1093/eurheartj/ehad152
- Aengevaeren VL, Mosterd A, Sharma S, et al. Exercise and Coronary Atherosclerosis: Observations, Explanations, Relevance, and Clinical Management. Circulation. 2020;141(16):1338-1350. doi:10.1161/CIRCULATIONAHA.119.044467
- Merghani A, Maestrini V, Rosmini S, et al. Prevalence of Subclinical Coronary Artery Disease in Masters Endurance Athletes With a Low Atherosclerotic Risk Profile. Circulation. 2017;136(2):126-137. doi:10.1161/CIRCULATIONAHA.116.026964
- O’Driscoll J, Parry-Williams G, Moser J, et al. Resting and exercise-induced occult hypertension and coronary atherosclerosis in male masters endurance athletes. Br J Sports Med. Published online May 14, 2026. doi:10.1136/bjsports-2025-111347
- Heidbuchel H, Adami PE, Antz M, et al. Recommendations for participation in leisure-time physical activity and competitive sports in patients with arrhythmias and potentially arrhythmogenic conditions: Part 1: Supraventricular arrhythmias. A position statement of the Section of Sports Cardiology and Exercise from the European Association of Preventive Cardiology (EAPC) and the European Heart Rhythm Association (EHRA), both associations of the European Society of Cardiology. Eur J Prev Cardiol. 2021;28(14):1539-1551. doi:10.1177/2047487320925635
- Sharma S, Drezner JA, Baggish A, et al. International recommendations for electrocardiographic interpretation in athletes. Eur Heart J. 2018;39(16):1466-1480. doi:10.1093/eurheartj/ehw631
- Cohn PF, Fox KM, Daly C. Silent myocardial ischemia. Circulation. 2003;108(10):1263-1277. doi:10.1161/01.CIR.0000088001.59265.EE
- Deedwania PC, Carbajal EV. Silent ischemia during daily life is an independent predictor of mortality in stable angina. Circulation. 1990;81(3):748-756. doi:10.1161/01.cir.81.3.748
- Katzel LI, Fleg JL, Busby-Whitehead MJ, et al. Exercise-induced silent myocardial ischemia in master athletes. Am J Cardiol. 1998;81(3):261-265. doi:10.1016/s0002-9149(97)00898-9
- Sheps DS, Maixner W, Hinderliter AL. Mechanisms of pain perception in patients with silent myocardial ischemia. Am Heart J. 1990;119(4):983-987. doi:10.1016/s0002-8703(05)80351-5
- Koltyn KF. Analgesia following exercise: a review. Sports Med. 2000;29(2):85-98. doi:10.2165/00007256-200029020-00002
- Naugle KM, Fillingim RB, Riley JL 3rd. A meta-analytic review of the hypoalgesic effects of exercise. J Pain. 2012;13(12):1139-1150. doi:10.1016/j.jpain.2012.09.006
- Heaps CL, Parker JL. Effects of exercise training on coronary collateralization and control of collateral resistance. J Appl Physiol (1985). 2011;111(2):587-598. doi:10.1152/japplphysiol.00338.2011
- Kannel WB, Abbott RD. Incidence and prognosis of unrecognized myocardial infarction. An update on the Framingham study. N Engl J Med. 1984;311(18):1144-1147. doi:10.1056/NEJM198411013111802
- Zhang ZM, Rautaharju PM, Prineas RJ, et al. Race and Sex Differences in the Incidence and Prognostic Significance of Silent Myocardial Infarction in the Atherosclerosis Risk in Communities (ARIC) Study. Circulation. 2016;133(22):2141-2148. doi:10.1161/CIRCULATIONAHA.115.021177
- Schelbert EB, Cao JJ, Sigurdsson S, et al. Prevalence and prognosis of unrecognized myocardial infarction determined by cardiac magnetic resonance in older adults. JAMA. 2012;308(9):890-896. doi:10.1001/2012.jama.11089
- Sheifer SE, Manolio TA, Gersh BJ. Unrecognized myocardial infarction. Ann Intern Med. 2001;135(9):801-811. doi:10.7326/0003-4819-135-9-200111060-00010
- Acharya T, Aspelund T, Jonasson TF, et al. Association of Unrecognized Myocardial Infarction With Long-term Outcomes in Community-Dwelling Older Adults: The ICELAND MI Study. JAMA Cardiol. 2018;3(11):1101-1106. doi:10.1001/jamacardio.2018.3285
- Turkbey EB, Nacif MS, Guo M, et al. Prevalence and Correlates of Myocardial Scar in a US Cohort. JAMA. 2015;314(18):1945-1954. doi:10.1001/jama.2015.14849
- Sheps DS, Adams KF, Hinderliter A, et al. Endorphins are related to pain perception in coronary artery disease. Am J Cardiol. 1987;59(6):523-527. doi:10.1016/0002-9149(87)91161-1
- Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74(5):1124-1136. doi:10.1161/01.cir.74.5.1124
- Małek ŁA, Miłosz-Wieczorek B, Marczak M. Diagnostic Yield of Cardiac Magnetic Resonance in Athletes with and without Features of the Athlete’s Heart and Suspected Structural Heart Disease. Int J Environ Res Public Health. 2022;19(8):4829. Published 2022 Apr 15. doi:10.3390/ijerph19084829
- Möhlenkamp S, Leineweber K, Lehmann N, et al. Coronary atherosclerosis burden, but not transient troponin elevation, predicts long-term outcome in recreational marathon runners. Basic Res Cardiol. 2014;109(1):391. doi:10.1007/s00395-013-0391-8
- Breuckmann F, Möhlenkamp S, Nassenstein K, et al. Myocardial late gadolinium enhancement: prevalence, pattern, and prognostic relevance in marathon runners. Radiology. 2009;251(1):50-57. doi:10.1148/radiol.2511081118
- van de Sande DAJP, Barneveld PC, Hoogsteen J, Doevendans PA, Kemps HMC. Coronary microvascular function in athletes with abnormal exercise test results. Neth Heart J. 2019;27(12):621-628. doi:10.1007/s12471-019-01336-6
- Camici PG, Crea F. Coronary microvascular dysfunction. N Engl J Med. 2007;356(8):830-840. doi:10.1056/NEJMra061889
- Maron DJ, Hochman JS, Reynolds HR, et al. Initial Invasive or Conservative Strategy for Stable Coronary Disease. N Engl J Med. 2020;382(15):1395-1407. doi:10.1056/NEJMoa1915922
- Reynolds HR, Shaw LJ, Min JK, et al. Outcomes in the ISCHEMIA Trial Based on Coronary Artery Disease and Ischemia Severity. Circulation. 2021;144(13):1024-1038. doi:10.1161/CIRCULATIONAHA.120.049755
- Mandsager K, Harb S, Cremer P, Phelan D, Nissen SE, Jaber W. Association of Cardiorespiratory Fitness With Long-term Mortality Among Adults Undergoing Exercise Treadmill Testing. JAMA Netw Open. 2018;1(6):e183605. Published 2018 Oct 5. doi:10.1001/jamanetworkopen.2018.3605
- Knuuti J, Ballo H, Juarez-Orozco LE, et al. The performance of non-invasive tests to rule-in and rule-out significant coronary artery stenosis in patients with stable angina: a meta-analysis focused on post-test disease probability. Eur Heart J. 2018;39(35):3322-3330. doi:10.1093/eurheartj/ehy267
- Knuuti J, Wijns W, Saraste A, et al. 2019 ESC Guidelines for the diagnosis and management of chronic coronary syndromes. Eur Heart J. 2020;41(3):407-477. doi:10.1093/eurheartj/ehz425
- Maron BJ, Zipes DP, Kovacs RJ. Eligibility and Disqualification Recommendations for Competitive Athletes With Cardiovascular Abnormalities: Preamble, Principles, and General Considerations: A Scientific Statement From the American Heart Association and American College of Cardiology. J Am Coll Cardiol. 2015;66(21):2343-2349. doi:10.1016/j.jacc.2015.09.032
- Pelliccia A, Sharma S, Gati S, et al. 2020 ESC Guidelines on sports cardiology and exercise in patients with cardiovascular disease. Eur Heart J. 2021;42(1):17-96. doi:10.1093/eurheartj/ehaa605
- Parry-Williams G, Sharma S. The effects of endurance exercise on the heart: panacea or poison?. Nat Rev Cardiol. 2020;17(7):402-412. doi:10.1038/s41569-020-0354-3
- Tsimikas S. A Test in Context: Lipoprotein(a): Diagnosis, Prognosis, Controversies, and Emerging Therapies. J Am Coll Cardiol. 2017;69(6):692-711. doi:10.1016/j.jacc.2016.11.042
- Sniderman AD, Thanassoulis G, Glavinovic T, et al. Apolipoprotein B Particles and Cardiovascular Disease: A Narrative Review. JAMA Cardiol. 2019;4(12):1287-1295. doi:10.1001/jamacardio.2019.3780
- Seiler S. What is best practice for training intensity and duration distribution in endurance athletes?. Int J Sports Physiol Perform. 2010;5(3):276-291. doi:10.1123/ijspp.5.3.276
- Stöggl T, Sperlich B. Polarized training has greater impact on key endurance variables than threshold, high intensity, or high volume training. Front Physiol. 2014;5:33. Published 2014 Feb 4. doi:10.3389/fphys.2014.00033
- Eijsvogels TMH, Thompson PD, Franklin BA. The “Extreme Exercise Hypothesis”: Recent Findings and Cardiovascular Health Implications. Curr Treat Options Cardiovasc Med. 2018;20(10):84. Published 2018 Aug 28. doi:10.1007/s11936-018-0674-3
- Eijsvogels TMH, Thompson PD, Franklin BA. The “Extreme Exercise Hypothesis”: Recent Findings and Cardiovascular Health Implications. Curr Treat Options Cardiovasc Med. 2018;20(10):84. Published 2018 Aug 28. doi:10.1007/s11936-018-0674-3