Molecular Traffic and Arterial Disease
How ApoB Lipoproteins and Rho GEF Signaling Help Explain Atherosclerosis
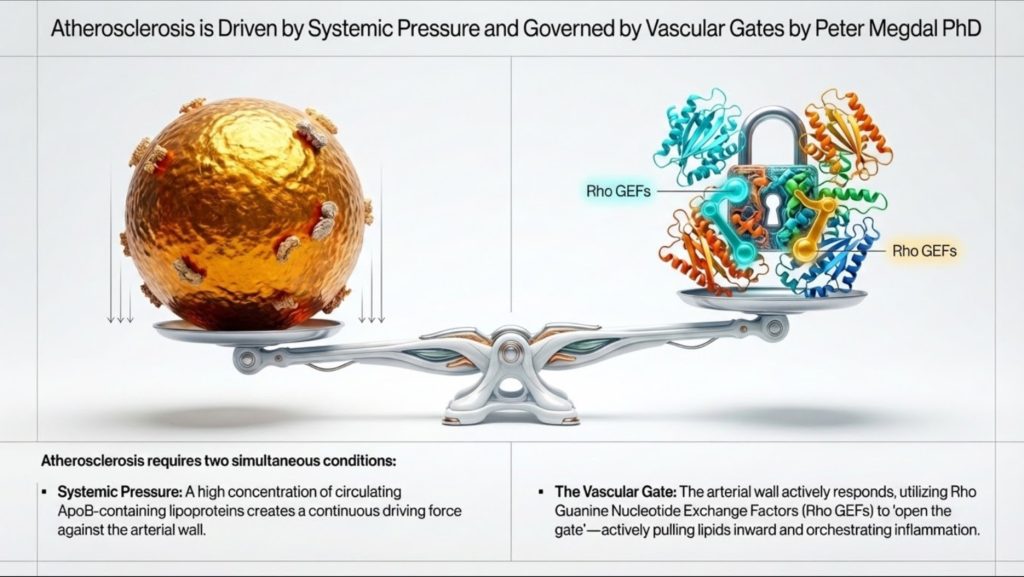
Atherosclerosis is often explained too simply. Most people are told that “high cholesterol” clogs arteries, and while that idea points in the right direction, it leaves out the deeper biology that actually drives disease. Cholesterol itself is not the whole story. The more accurate picture is that atherosclerosis develops when atherogenic lipoprotein particles enter the artery wall, become trapped, and trigger a long inflammatory cascade. Whether that happens depends not only on how many particles are circulating in the blood, but also on how the arterial wall behaves at the molecular level [1-8].
That second part matters more than most discussions acknowledge. The arterial wall is not a passive pipe. It is an active, responsive tissue. Endothelial cells regulate which particles cross into the vessel wall. Immune cells decide how to react once those particles are retained. Smooth muscle cells then help determine whether a plaque stabilizes or becomes dangerous. Much of this local regulation is controlled by a family of molecular switches known as Rho guanine nucleotide exchange factors, or Rho GEFs, working through Rho GTPases such as RhoA, Rac1, and Cdc42 [4].
This provides a more complete way to think about cardiovascular disease. Systemic lipid burden, especially the number of ApoB-containing particles, sets the pressure in the system. But local signaling inside the arterial wall determines whether those particles are allowed in, retained, and transformed into plaque. In practical terms, ApoB measures the number of vehicles on the road, while Rho GEF signaling helps decide how open the gate is and how destructive the local response will be [1-4].
Understanding this relationship helps bridge high-level clinical lipidology and modern vascular biology. It also makes the disease easier to explain to the public. Atherosclerosis is not just “fat in the blood.” It is a problem of particle traffic, arterial entry, retention, immune activation, and tissue remodeling.
Why Lipoproteins Exist in the First Place
The human body has a basic engineering problem. It needs to transport lipids such as triglycerides and cholesterol, but blood plasma is mostly water. Lipids are hydrophobic, meaning they do not dissolve well in an aqueous environment. If the body simply released raw fat into the bloodstream, it would separate and clump, much like oil in water [1,2].
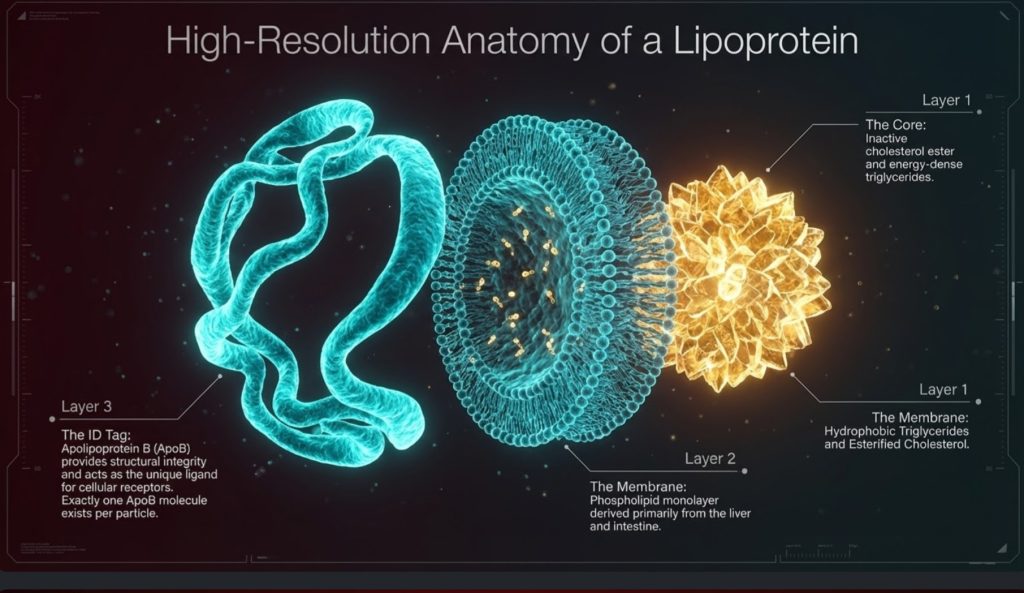
The solution is the lipoprotein. Lipoproteins are transport particles built to carry hydrophobic cargo through watery plasma. They have a hydrophobic core, where triglycerides and cholesterol esters are stored, and a more water-compatible surface made of phospholipids, free cholesterol, and proteins called apolipoproteins [1,2,5].
This is important because lipoproteins are often treated as abstract lab values, but they are better understood as physical transport vehicles. They move energy and structural materials through the body. Triglycerides are the main energy cargo. Cholesterol provides structural support for membranes, steroid hormone synthesis, and bile acid production. The bloodstream is the highway, and lipoproteins are the fleet [1-3,5].
From that viewpoint, the body did not evolve lipoproteins mainly to move cholesterol. It evolved them to move energy. Cholesterol is necessary, but the transport system is deeply tied to fuel delivery. That distinction matters, because it helps clarify one of the most misunderstood issues in cardiovascular disease: the body does need cholesterol, but it does not need large quantities of cholesterol moving through plasma to keep tissues alive and functioning [2,11-15].
Cholesterol Homeostasis: How Little Is Actually Needed
A central but often overlooked fact in lipid biology is that the body requires only a very small amount of cholesterol at the cellular level. Every cell membrane needs cholesterol for structure and fluidity, and certain specialized tissues need it for steroid hormone synthesis. But that requirement is remarkably small, tightly regulated, and mostly satisfied locally rather than through bulk delivery from circulating LDL [2,11-17].
Cholesterol homeostasis is governed by highly regulated intracellular signaling pathways that maintain sufficiency while preventing toxicity. Contrary to common assumptions, virtually all nucleated cells possess the ability to synthesize their own cholesterol. They do not simply wait for lipoproteins to deliver it. They are genetically equipped to make what they need [11,12].
The central regulatory system is the SREBP-2–SCAP–INSIG axis. When intracellular cholesterol levels fall, sterol regulatory element-binding protein 2 (SREBP-2) is escorted by SREBP cleavage-activating protein (SCAP) from the endoplasmic reticulum to the Golgi apparatus. There, SREBP-2 undergoes proteolytic activation. The active transcription factor then enters the nucleus and upregulates genes involved in cholesterol synthesis and uptake, including HMG-CoA reductase (HMGCR) and the LDL receptor (LDLR) [11,12].
This drives the mevalonate pathway, beginning with acetyl-CoA and proceeding through HMG-CoA and mevalonate toward cholesterol synthesis. Because this machinery exists throughout the body, cells do not depend on large plasma pools of cholesterol to preserve membrane integrity or basic survival [11,12].
When intracellular cholesterol rises, the system shuts itself down. Cholesterol and oxysterols promote INSIG-mediated retention of the SCAP–SREBP complex in the endoplasmic reticulum, reducing further transcriptional activation. At the same time, HMG-CoA reductase is targeted for degradation through ubiquitin-proteasome pathways. In other words, cholesterol metabolism is controlled by a built-in feedback loop designed to avoid excess accumulation [11,12].
That excess matters, because free cholesterol is not benign when it builds up inside cells. Too much intracellular cholesterol can destabilize membranes, crystallize in lysosomes, trigger inflammatory signaling such as the NLRP3 inflammasome, and promote apoptosis or necrosis [13,14]. This is one reason the body is not designed to stockpile it.
Cells therefore rely not only on synthesis and feedback inhibition, but also on active export. Through Liver X Receptor (LXR) signaling, oxysterols induce transporters such as ABCA1 and ABCG1, which move excess cholesterol out of cells and toward HDL-mediated reverse cholesterol transport [15].
Taken together, these pathways show something important: cells need cholesterol, but only in small, controlled amounts. The dominant biological problem is not acquiring more cholesterol. It is balancing synthesis, use, storage, and efflux so that excess does not become toxic [11-15].

Tissue-Specific Cholesterol Independence
This principle becomes even clearer when looking at specific organs. Many tissues synthesize cholesterol locally and do not rely heavily on circulating LDL.
The central nervous system is the most striking example. Because apoB-containing lipoproteins do not meaningfully cross the blood-brain barrier, the brain must largely produce its own cholesterol. Astrocytes synthesize cholesterol and distribute it locally through apoE-containing lipoproteins, a process linked to LXR-regulated pathways [16].
The same is broadly true of steroidogenic tissues, including the adrenal glands and gonads. These tissues can synthesize cholesterol de novo from acetate. Although they express receptors such as LDLR and SR-B1, their dependence on circulating LDL under ordinary conditions appears limited, with uptake becoming more relevant in specialized or high-demand states such as acute ACTH stimulation [2,17].
This supports an important physiological point. The body is not dependent on high circulating LDL-C levels for normal endocrine or cellular function. Indeed, mechanistic and clinical discussions in lipidology have emphasized that very low LDL-C levels, even around 10 mg/dL, do not necessarily imply impaired steroidogenesis or membrane failure when intracellular synthesis remains intact [2,11,17].
So while cholesterol is essential, the amount actually required in circulation for cellular survival is far smaller than many people assume. That reality reinforces the view that elevated circulating ApoB particles are not a biological necessity; they are a risk exposure.
The ApoB Fleet: Why Particle Number Matters
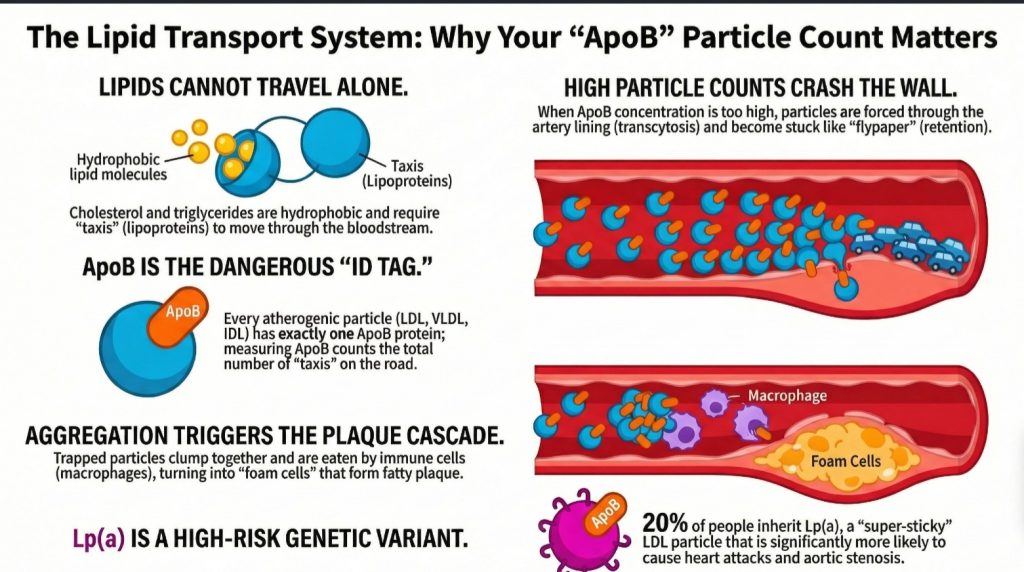
Among the many lipoproteins in circulation, the most relevant to atherosclerosis are the ApoB-containing particles. These include chylomicrons and their remnants, very low-density lipoproteins (VLDL), intermediate-density lipoproteins (IDL), low-density lipoproteins (LDL), and lipoprotein(a), or Lp(a) [1,2].
The key clinical insight is that each atherogenic particle contains exactly one ApoB molecule. That means ApoB serves as a direct count of the number of atherogenic particles in circulation. By contrast, LDL-C measures only the mass of cholesterol contained within LDL particles. A person can have a relatively normal LDL-C but still have a high number of particles if those particles are small and cholesterol-depleted. In that setting, risk can be underestimated if clinicians look only at LDL-C [1-3].
This is why many lipid experts now view ApoB as the more biologically meaningful metric. It reflects how many opportunities there are for particles to enter the artery wall. Put simply, if every ApoB particle is one potential arterial collision, then ApoB tells us the traffic density [1-3].
That traffic analogy becomes especially useful when explaining risk to non-specialists. The issue is not just how much cargo is in each truck. The issue is how many trucks are on the road, how long they stay there, and how likely they are to crash into the side wall.
This is also where the minimal-cholesterol discussion becomes clinically relevant. If tissues can synthesize the modest cholesterol they need, then high ApoB counts are not best understood as a beneficial delivery reserve. They are better understood as an increased burden of potentially arterial-penetrating particles [2,11-17].
From Fuel Delivery to LDL Persistence
The story begins with triglyceride-rich particles. The intestine produces chylomicrons to transport dietary fat. The liver produces VLDL to transport endogenous triglycerides. These are large particles whose major job is fuel delivery [1,2].
As these particles circulate, they interact with lipoprotein lipase (LPL), an enzyme that unloads triglycerides for use by muscle, heart, and adipose tissue. As triglycerides are removed, the particle shrinks. Surface material is shed. But the ApoB backbone remains. Over time, VLDL is remodeled into IDL and then LDL [1,2,18].
LDL is therefore not some rogue or evil particle by design. It is a downstream product of normal lipoprotein metabolism. What makes LDL especially important is its persistence. LDL stays in circulation much longer than larger triglyceride-rich particles, often for 2 to 5 days, which increases the statistical chance that it will cross into the arterial wall [1,2].
This is where the disease model starts to sharpen. Atherogenesis is not caused simply because LDL exists. It is caused because enough ApoB particles remain in circulation long enough to enter the intima and become retained.
At the same time, this reinforces an important conceptual distinction. The presence of LDL in blood is not proof that tissues are desperate for cholesterol. Much of LDL biology reflects the remodeling and persistence of ApoB particles after triglyceride transport. In that sense, LDL is not merely a “cholesterol delivery truck.” It is also a long-circulating remnant of systemic lipid transport with increased opportunity to interact with the vessel wall [1,2,18].
Atherosclerosis Begins With Entry, Not Just Presence
One of the most useful upgrades in modern cardiovascular thinking is the realization that atherosclerosis does not begin merely because particles are in the blood. It begins when they cross the endothelial barrier and lodge in the subendothelial space, or intima [1,6-8].
That means the artery wall matters enormously. The endothelium is only one cell layer thick, but it is biologically active. It senses blood flow, responds to inflammatory signals, regulates barrier function, and controls which materials pass through. Certain arterial regions, especially branch points and curvatures, are more prone to plaque formation because the local flow pattern is disturbed. Those disturbed flow zones create a molecular environment that favors atherogenesis [4,6-8].
Historically, many people imagined LDL simply leaking through a damaged endothelium. The newer picture is more precise: LDL entry is an active, regulated process called transcytosis [6-8].
This matters conceptually. If particle entry is regulated, then atherosclerosis is not just about what is in plasma. It is also about how the arterial wall responds to that plasma environment. Systemic exposure and local permissiveness work together.
DOCK4, SR-B1, and Regulated LDL Transcytosis
One of the most intriguing findings in recent years is that endothelial cells can actively transport LDL across themselves through a mechanism involving SR-B1 and DOCK4 [6-8].
SR-B1, or scavenger receptor class B type 1, has long been discussed in the context of HDL biology, but it also appears to play a role in the uptake and transcytosis of LDL under certain conditions. DOCK4 is a Rho GEF that activates Rac1, which helps drive the cytoskeletal changes and vesicular movement needed for this transport process [4,7,8].
This matters because it reframes the earliest step of atherogenesis. LDL entry into the vessel wall is not just accidental seepage. It is at least partly controlled by a specific signaling system. Even more striking, DOCK4 expression has been observed to increase in atherosclerosis-prone regions before visible lesions are present, suggesting that the artery wall may become permissive long before disease is obvious on imaging or pathology [4,8].
Clinically, that opens a provocative possibility. ApoB tells us how many particles are available to enter. But the DOCK4/SR-B1/Rac1 system may help determine how easily they can do so. In other words, ApoB measures the pressure at the gate; DOCK4 helps regulate the gate itself.
Retention: The Point of No Return
Entry alone is not enough. Once an ApoB particle reaches the intima, the next key event is retention. These particles bind to matrix molecules called proteoglycans, which act almost like arterial flypaper [1,3].
This step is crucial because a retained particle is now exposed to a very different microenvironment. Inside the arterial wall, it can undergo a series of modifications. It may oxidize, but oxidation is only part of the story. Lipases and other enzymes can alter the particle surface, generating lipids such as sphingomyelin and ceramide that make particles more adhesive and more likely to aggregate [4].
That aggregation step is especially important. A single isolated retained LDL particle is not the same thing biologically as a sticky cluster of modified particles. Once particles aggregate, they become far more likely to provoke macrophage uptake and foam cell formation [1,4].
For clinicians and informed readers alike, this is a useful distinction: the danger is not just circulating LDL or even intimal LDL. The danger is retained, modified, aggregated ApoB-containing lipoprotein.
This is also the point at which “cholesterol toxicity” becomes context-specific. Cholesterol is necessary in tiny, regulated intracellular amounts, but once ApoB particles are trapped and modified in the arterial wall, their lipid cargo becomes part of a highly toxic inflammatory environment. The same sterol that is essential in membranes can become pathologic when delivered into the wrong tissue compartment in the wrong form [13,14].
Leukocyte Recruitment: How the Artery Becomes Inflamed
Once the intima contains retained and modified lipoproteins, the lesion shifts from a lipid problem to an inflammatory one. At this stage, the recruitment of monocytes and other leukocytes becomes central [4].
A Rho GEF called SGEF, also known as Arhgef26, appears to play a major role here. SGEF helps endothelial cells form ICAM-1-dependent docking structures, actin-rich protrusions that allow leukocytes to adhere more effectively under flow conditions. These structures are especially relevant in the high-shear environment of arteries, where immune cells might otherwise be washed away [4].
Another related signaling molecule, Arhgef1, links inflammatory recruitment to Angiotensin II signaling. This creates a mechanistic bridge between hypertension and arterial inflammation. High blood pressure is not merely a hemodynamic burden; it can also amplify leukocyte recruitment through RhoA-mediated pathways [4].
This helps explain why cardiovascular risk factors often cluster biologically. Hypertension, insulin resistance, and high ApoB do not act in isolation. They converge within the arterial wall, where endothelial signaling, immune recruitment, and lipoprotein retention reinforce one another.
Foam Cells and the Macrophage Response
Macrophages are often described as the body’s cleanup crew. In atherosclerosis, however, the cleanup operation can become self-destructive. Macrophages ingest modified and aggregated lipoproteins, become loaded with cholesterol, and transform into foam cells [1,4].
This step is not passive. It depends heavily on cytoskeletal remodeling and receptor signaling. The Vav family of Rho GEFs, especially Vav1, Vav2, and Vav3, helps regulate these processes. Vav proteins influence CD36-mediated uptake of oxidized lipoproteins and support formation of the lysosomal synapse, which allows macrophages to interact with and digest large lipoprotein aggregates [4].
As foam cells accumulate, they release inflammatory mediators, eventually die, and contribute to the necrotic core of the plaque. This transforms a microscopic lesion into a clinically meaningful plaque. Over time, the lesion can calcify, remain stable, or become vulnerable to rupture [1,3].
This is another point where the molecular framing adds value. The disease is not just about “too much cholesterol.” It is about how immune cells process abnormal lipid material inside a specific tissue environment.
The added cholesterol-homeostasis literature deepens this picture. Foam cell formation is not merely an issue of passive lipid storage. It is a breakdown of normal cholesterol handling. When macrophage uptake overwhelms intracellular control and efflux systems, free cholesterol accumulates, inflammatory pathways are activated, and cell death follows. In that sense, plaque progression can be understood as a localized failure of cholesterol homeostasis inside the arterial wall [13-15].
Smooth Muscle Cells and Plaque Stability
As plaques mature, vascular smooth muscle cells (VSMCs) become major players. These cells normally help maintain vascular tone, but in atherosclerosis they can change phenotype. They may migrate from the media to the intima, proliferate, and contribute to plaque structure [4].
This behavior is influenced by Rho GEF signaling. Molecules such as Kalirin and Vav3 promote migration and proliferation through Rac1- and RhoA-related pathways. Some of this remodeling may be protective, because VSMCs can help build a fibrous cap over the plaque. But excessive or disordered remodeling can also contribute to luminal narrowing and plaque instability [4].
By contrast, Arhgef7, also called beta-PIX, appears to support barrier integrity and protective cell polarity pathways. This suggests that plaque biology is not just driven by pro-disease signals. It is also shaped by counter-regulatory systems trying to preserve arterial structure [4].
The stability of a plaque therefore depends on a balance between damage, repair, inflammation, and remodeling. That balance is partly governed by the same family of molecular switches that influenced the lesion from the beginning.
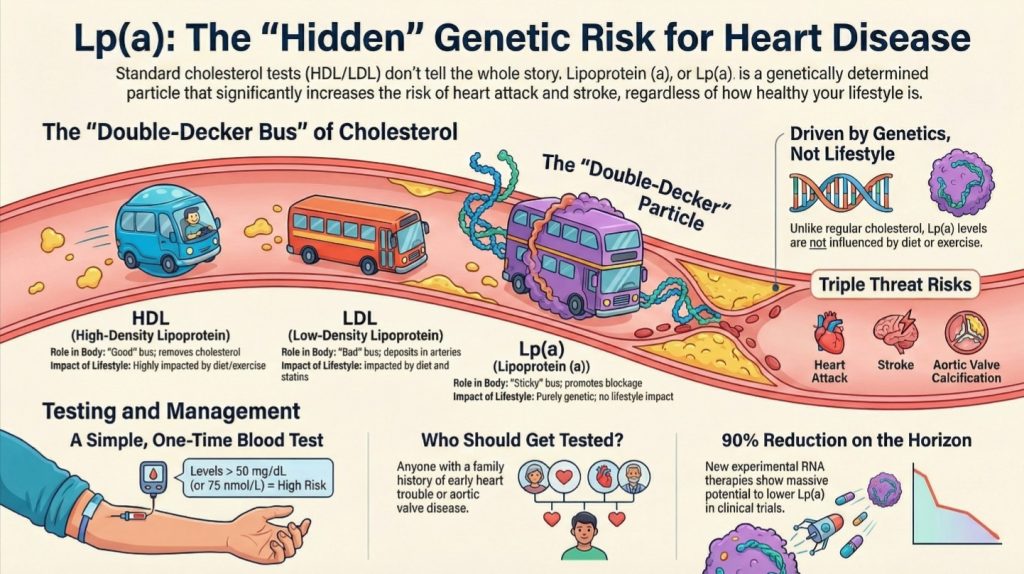
Lp(a): The Genetic Risk Multiplier
No modern discussion of lipid-driven cardiovascular disease is complete without lipoprotein(a), or Lp(a). Structurally, Lp(a) is an LDL-like particle with an additional apolipoprotein(a) attached. Clinically, it is one of the most important inherited cardiovascular risk factors [1-3,5,9].
About one in five people has an elevated Lp(a), and levels are largely genetically determined. Unlike standard LDL, Lp(a) is not significantly improved by diet or exercise and is only modestly affected by most conventional lipid therapies [1-3,5,9].
Lp(a) appears to be especially dangerous because it is not only atherogenic but also pro-inflammatory and pro-thrombotic. Elevated levels are linked to premature myocardial infarction, stroke, and aortic valve stenosis. For this reason, many experts now recommend that every patient have Lp(a) measured at least once in a lifetime [1-3,5,9].
This is a major public health message. Someone can do many things right and still carry substantial inherited risk through elevated Lp(a). Identifying that risk early matters.
Why ApoB Is a Better Clinical Compass Than LDL-C
For everyday practice, the main diagnostic lesson is simple: ApoB is closer to the biology of disease than LDL-C [1-3].
LDL-C estimates how much cholesterol mass is present in LDL particles. ApoB estimates how many atherogenic particles are circulating. Since each particle is one potential entrant into the artery wall, ApoB better captures the opportunity for arterial injury.
Non-HDL cholesterol is often a better surrogate than LDL-C, because it includes the cholesterol carried by all ApoB-containing particles. But ApoB remains the cleaner particle-based measure. Coronary artery calcium scoring can help detect established disease later in life, and Lp(a) helps identify inherited risk. But for understanding the causal burden of circulating atherogenic particles, ApoB is central [1-3].
This does not mean LDL-C is useless. It remains clinically valuable and widely available. But when the goal is to align testing with mechanism, ApoB is the stronger marker.
It also fits more cleanly with the physiology described above. If tissues do not require large amounts of circulating cholesterol to survive, and if cholesterol needs are largely met by local synthesis and tightly regulated intracellular pathways, then the clinically important question becomes not “How much cholesterol is present in plasma?” but “How many atherogenic particles are repeatedly interacting with the artery wall?” ApoB answers that better than LDL-C [2,11-17].
Therapeutic Implications: Today and Tomorrow
Current lipid-lowering therapy still matters enormously. Statins, ezetimibe, and PCSK9 inhibitors reduce atherogenic particle burden and have strong outcome data. PCSK9 inhibitors also lower Lp(a) modestly, and newer RNA-based therapies targeting Lp(a) are showing dramatic reductions in trials [1,3,9].
Lifestyle remains foundational. Weight loss, improved insulin sensitivity, reduced hepatic VLDL production, and lower inflammatory burden all help reduce systemic risk [1]. But the emerging biology suggests that the future of cardiovascular prevention may not stop at lowering plasma particles. It may also involve targeting the arterial wall directly.
That is where Rho GEF biology becomes especially exciting. If specific GEFs such as DOCK4 or SGEF are key regulators of LDL entry or leukocyte recruitment, then direct inhibition of those pathways might offer a more selective way to interfere with plaque formation [4]. Instead of only reducing traffic on the road, clinicians might one day also tighten the gate and blunt the inflammatory response inside the vessel wall.
This is still an emerging area, not a routine clinical strategy. But conceptually it points toward precision medicine: targeting the specific local pathways that convert circulating risk into arterial disease.
The integrated cholesterol-homeostasis framework also helps explain why aggressive lowering of ApoB burden is biologically plausible. If intracellular needs can still be met through endogenous synthesis and tightly regulated tissue handling, then reducing circulating atherogenic particles need not imply cellular deprivation. It may simply reduce unnecessary arterial exposure [2,11-17].
A Better Way to Explain Atherosclerosis
For the general public, the disease can be summarized in a way that is both intuitive and accurate.
ApoB-containing lipoproteins are like transport trucks carrying fuel and structural cargo through the bloodstream. If there are too many trucks on the road, some eventually cross into the artery wall. Once trapped there, they become damaged and sticky. The immune system sends in cleanup cells, but those cells get overloaded and create plaque. Meanwhile, the artery wall itself is not passive. It has molecular gatekeepers that regulate particle entry, immune docking, cholesterol handling, and tissue remodeling [1-8,11-15].
That framework is simple enough for public education but detailed enough to be useful to clinicians. It avoids the misleading oversimplification that cholesterol alone “clogs” arteries while preserving the core truth that cholesterol-containing ApoB particles are causally necessary for disease [1-3].
It also leaves room for an important nuance: the body needs cholesterol, but only in very small, tightly controlled amounts. The real danger is not the existence of cholesterol itself. The danger is the chronic circulation, arterial entry, and retention of ApoB-containing particles in a tissue environment that turns their cargo into inflammation and plaque.
Conclusion
Atherosclerosis is best understood as the intersection of two systems. The first is systemic lipoprotein biology, where ApoB-containing particles circulate as the necessary carriers of triglycerides and cholesterol. The second is local arterial biology, where endothelial cells, leukocytes, macrophages, and smooth muscle cells decide whether those particles become plaque.
ApoB captures the number of circulating atherogenic particles and therefore the statistical opportunity for arterial injury. Rho GEF–Rho GTPase signaling helps explain why and how that injury occurs within the vessel wall. Together, these systems offer a richer model of disease than the old LDL-C-only framework.
At the same time, cholesterol itself is governed by a highly regulated intracellular economy. Through the SREBP-2–SCAP–INSIG system, the mevalonate pathway, LXR-mediated efflux signaling, and tissue-specific synthesis, the body maintains the small amounts of cholesterol it needs while defending itself against excess [11-17]. This means the physiological requirement for cholesterol is real, but the amount required is modest, locally managed, and fundamentally different from the pathological burden imposed by excess circulating ApoB particles.
For clinicians, this supports a stronger emphasis on ApoB, Lp(a), and early prevention. For the public, it clarifies that cardiovascular disease is not simply about eating fat or having “high cholesterol.” It is about how many atherogenic particles circulate, how long they persist, how the artery wall responds to them, and how failures of local cholesterol handling turn retained particles into inflammation and plaque.
The future of lipidology may therefore be dual: lowering the number of dangerous particles in the bloodstream while also targeting the molecular programs that let those particles enter and injure the artery wall in the first place [1-10]. In that broader framework, one of the most important conceptual corrections is this: the body does not need a large circulating surplus of cholesterol to survive. It needs only a very small, carefully regulated amount at the cellular level. Atherosclerosis begins when the transport system overshoots that need and the artery wall pays the price.
References
- Perone F, Bernardi M, Spadafora L, et al. Non-Traditional Cardiovascular Risk Factors: Tailored Assessment and Clinical Implications. J Cardiovasc Dev Dis. 2025;12(5):171. Published 2025 Apr 28. doi:10.3390/jcdd12050171
- Attia P, Dayspring T. #20 – Tom Dayspring, M.D., FACP, FNLA – Part I of V: an introduction to lipidology.
- Hill S. What Causes Cardiovascular Disease? Lipid Series Part 1. The Proof with Simon Hill.
- Li M, Jiao Q, Xin W, et al. The Emerging Role of Rho Guanine Nucleotide Exchange Factors in Cardiovascular Disorders: Insights Into Atherosclerosis: A Mini Review. Front Cardiovasc Med. 2022;8:782098. Published 2022 Jan 3. doi:10.3389/fcvm.2021.782098
- Heart Matters. Cholesterol Explained: HDL, LDL, and the Hidden Risk of Lp(a).
- Hackethal V. Research Suggests LDL Is Actively Transported Into Arterial Walls. NeurologyLive.
- Getz GS, Reardon CA. The “HDL receptor” scavenger receptor class B type 1 finesses the uptake of low-density lipoproteins into the subendothelial space of arteries. Biotarget.
- Huang L, Chambliss KL, Gao X, et al. SR-B1 drives endothelial cell LDL transcytosis via DOCK4 to promote atherosclerosis. Nature. 2019;569(7757):565-569. doi:10.1038/s41586-019-1140-4
- Nutrition Made Simple. Everything you need to know about Lp(a) | ft. Dr. Tom Dayspring.
- Heart Fit Clinic. Webinar on Cholesterol from a Lipidologist. Dr. Tom Dayspring.
- Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89(3):331-340. doi:10.1016/s0092-8674(00)80213-5
- Brown MS, Goldstein JL. Sterol regulatory element binding proteins (SREBPs): controllers of lipid synthesis and cellular uptake. Nutr Rev. 1998;56(2 Pt 2):S1-S75. doi:10.1111/j.1753-4887.1998.tb01680.x
- Muneer PMA, Alikunju S, Mishra V, et al. Activation of NLRP3 inflammasome by cholesterol crystals in alcohol consumption induces atherosclerotic lesions. Brain Behav Immun. 2017;62:291-305. doi:10.1016/j.bbi.2017.02.014
- Feng B, Yao PM, Li Y, et al. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat Cell Biol. 2003;5(9):781-792. doi:10.1038/ncb1035
- Murphy AJ, Akhtari M, Tolani S, et al. ApoE regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. J Clin Invest. 2011;121(10):4138-4149. doi:10.1172/JCI57559
- Dietschy JM, Turley SD. Cholesterol metabolism in the brain. Curr Opin Lipidol. 2001;12(2):105-112. doi:10.1097/00041433-200104000-00003
- Miller WL. MECHANISMS IN ENDOCRINOLOGY: Rare defects in adrenal steroidogenesis. Eur J Endocrinol. 2018;179(3):R125-R141. doi:10.1530/EJE-18-0279
- Ginsberg HN. Lipoprotein metabolism and its relationship to atherosclerosis. Med Clin North Am. 1994;78(1):1-20. doi:10.1016/s0025-7125(16)30174-2