
The 12-Year-Old with a 60-Year-Old’s Heart: The Surprising Science of the World’s Rarest Cholesterol Condition
1. Introduction: The Invisible Speed-Run
Imagine your body is a car, and your health is tracked by an odometer. For most of us, that odometer ticks up slowly and steadily. We might drive for 50 or 60 years before the engine starts to show significant wear and tear. But for some children, the odometer is spinning wildly out of control from the moment they are born. This is the reality of Homozygous Familial Hypercholesterolemia, or HoFH.
While most people think of heart disease as a “slow crawl” that happens in old age, for children with HoFH, it is a high-stakes “sprint” that actually begins before they are even out of the womb. To understand HoFH, you have to look at the liver. In a healthy body, the liver acts like a garbage collection system, constantly pulling “trash” (in the form of LDL cholesterol) out of the blood. In a child with HoFH, that garbage collection system is completely broken. Because the liver cannot clear the fat, it builds up to dangerous levels instantly. This turns what should be a lifelong journey into a medical emergency—a race against time where the heart is forced to age decades in just a few short years.
2. The “Speeding Odometer”: Understanding Cumulative Burden
To truly grasp why HoFH is so dangerous, we have to look past a single blood test and consider the Cumulative Cholesterol Burden. This is the most important concept in understanding why these children are in such danger. Think of cholesterol damage like rust forming inside a metal pipe. If only a little water flows through, the rust takes decades to build up. But if the water is filled with corrosive acid, that pipe will reach its breaking point in a heartbeat.
Medical science uses a specific measurement for this called mmol/L-years. This isn’t just a snapshot of how much cholesterol is in the blood today; it is a measure of the total “dose” of cholesterol the arteries have been forced to endure over a lifetime. It is the weight of the burden over time.
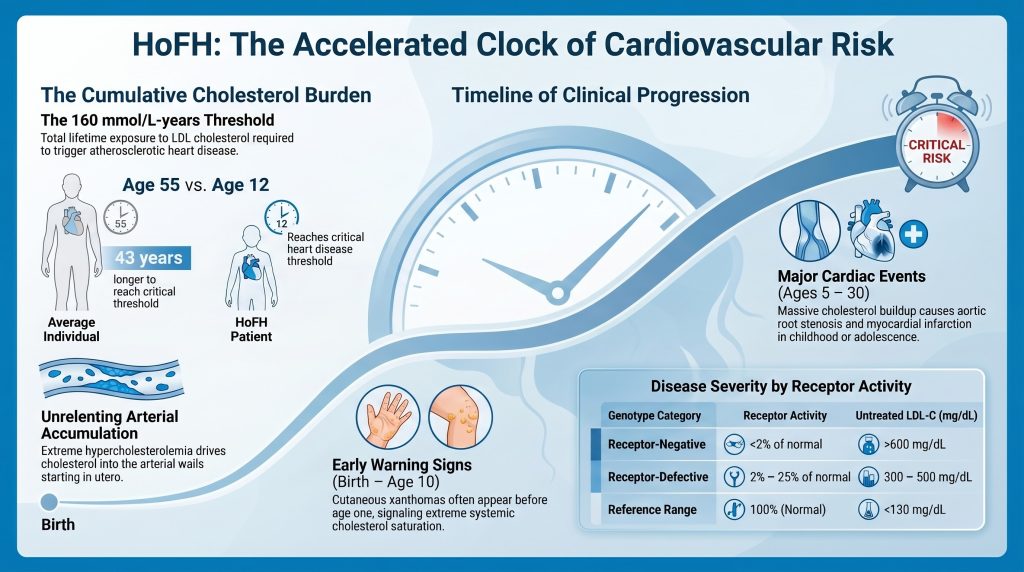
“The extreme cardiovascular risk in HoFH is driven by the unrelenting accumulation of cholesterol within the arterial wall, starting in utero.”
Research shows us that for an average person, the heart starts to develop significant disease—what doctors call Atherosclerotic Cardiovascular Disease (ASCVD)—when they hit a cumulative threshold of about 160 mmol/L-years. Most people don’t reach that “rust level” until they are 55 years old. However, a child with HoFH is born into a different reality. Because their levels are so high from day one, they can hit that 160 mark by the time they are just 12 years old. Even more frightening is the threshold for a first heart attack, which is approximately 125 mmol/L-years. An untreated child with the most severe form of this condition can reach that “heart attack zone” before they even finish elementary school.
3. When Your Body Wears Its Warning Signs: The Physical Clues
In many diseases, the danger is hidden deep inside. With HoFH, the body often tries to warn us by wearing its symptoms on the outside. When the blood is so saturated with fat that the vascular system can no longer contain it, the excess cholesterol begins to “spill over” into other tissues. This creates physical markers known as xanthomas.
To a parent or a doctor, these might look like strange yellow lumps or cholesterol bumps on a toddler’s skin. They are most commonly found in areas of friction, like the elbows, knees, buttocks, or the Achilles tendons. These are what doctors call pathognomonic signs—which is just a fancy way of saying they are “classic signals” that something is very wrong. Seeing these bumps on a child under the age of four is a “red alert.” It means the body’s “trash” is piling up on the sidewalk because the garbage trucks have stopped running.
We have known about these signs for over a century, and the history is heartbreaking. In 1889, doctors described two sisters with what we now know was HoFH. The 11-year-old sister had “egg-like tumors” on her hands and massive growths on her heels. She died shortly after a surgery, and the autopsy revealed a tragedy: her heart looked like that of an old man. Her Aorta—the body’s main blood highway—was thickened with fat, and her left carotid artery, which carries blood to the brain, was nearly completely blocked.
“Autopsy findings documented the aorta was thickened with fat-containing tissue and sclerotic plaques, while the left carotid was nearly completely occluded.”
This historical case shows that HoFH is not just a “cholesterol problem”—it is a total body saturation that targets the most vital pathways of life.
4. Blood That Looks Like Milk: The Extreme Magnitude
The sheer amount of cholesterol in an HoFH patient is almost impossible to imagine. Most doctors want your LDL cholesterol (the “bad” kind) to be under 130 mg/dL. In children with HoFH, those levels are regularly over 400, and in the most severe cases, they can soar above 1,000 mg/dL.
At these levels, the blood physically changes. The source context describes how the blood can take on a “visually distinct, opaque appearance” because the concentration of fat is so high. It essentially begins to look like milk or a thick cream rather than a translucent red liquid.
To visualize this, imagine a highway. In a normal person, there is a steady flow of traffic. The cars on this highway are ApoB particles. Think of ApoB as the “chassis” or the frame of every single “car” carrying cholesterol through your blood. In a healthy body, these cars exit the highway at the liver’s “off-ramps” (LDL receptors).
But in HoFH, there are 10 times as many cars on that same road, and the off-ramps are closed. This creates a permanent, non-stop traffic jam. Because the cars have nowhere to go, they eventually crash into the side walls of the highway—your arterial walls. This is especially dangerous at the aortic root and the coronary ostia (the “entrance ramps” to the heart’s own fuel lines). When fat builds up there, it causes Supravalvular Aortic Stenosis (SVAS), a dangerous narrowing right at the heart’s main exit valve. This isn’t just a clog; it’s a structural barrier made of fat.
5. The Genetic Lottery: Why Some Survive Longer
One of the most fascinating aspects of HoFH is its heterogeneity, or the way it varies from person to person. You might have two children with the same condition, yet one faces a crisis at age 5 while another lives into their 50s. This is the “Genetic Lottery.”
The severity depends on exactly how “broken” the liver’s garbage trucks are. Doctors look at the LDLR genotype:
- Receptor-negative: These patients have less than 2% of normal liver activity. They have essentially zero working garbage trucks. This is the most dangerous form, often leading to heart attacks in early childhood.
- Receptor-defective: These patients have between 2% and 25% activity. They have a few “broken” trucks that still manage to haul away some cholesterol, which can delay heart disease by a decade or more.
But there is an even more surprising factor: Protective Genetic Modifiers. Some people are born with “bonus” genes that act like a secret shield. For example, a “loss-of-function” variant in a gene called PCSK9 or ANGPTL3 can naturally lower cholesterol.
Consider the “Tokyo Case” from 1987—a 57-year-old man who survived for decades despite having HoFH. He had a “Class 4” mutation, an internalization defect. This meant his liver’s garbage trucks could “grab” the trash but couldn’t pull it inside the garage. It was enough to keep his levels at 461 mg/dL—still very high, but low enough to allow him to reach his 50s. More recently, a 59-year-old Czech patient was discovered who survived decades without even being diagnosed. These “lucky” survivors are the heroes of science; by studying their “secret shields,” researchers have learned how to create new medicines for everyone else.
6. The End of the “Death Sentence”: The Modern Treatment Era
For most of history, an HoFH diagnosis was a death sentence. Without treatment, the average age of death was just 18 years old. But we are now entering a “Modern Treatment Era” that is fundamentally changing the “clinical trajectory” of the disease. We are moving from watching the odometer spin to finally hitting the brakes.
The breakthrough has come from drugs that don’t rely on the broken liver receptors. If the main “highway exit” is closed, these drugs find a “back alley” or a “secret tunnel.”
- Lomitapide: This drug stops the liver from even building the “cars” (VLDL/LDL) in the first place.
- Evinacumab: This medicine targets the ANGPTL3 pathway, clearing cholesterol out of the blood through a system that doesn’t need the standard LDL receptor.
We know these treatments work because of data from the CASCADE FH Registry. The registry shows a stark difference between generations. Children diagnosed early—at a median age of 2.5 years—and started on aggressive, “receptor-independent” therapies are now reaching adulthood with much healthier hearts. In the past, people were often not diagnosed until age 23, after the damage was already done.
“Contemporary registry data demonstrate that early diagnosis and the use of advanced, receptor-independent therapies are successfully bending the clinical trajectory away from early mortality.”
7. Conclusion: A Race Against the Clock
HoFH is a medical emergency that starts before a child takes their first breath. It is a condition where the “cumulative burden” of cholesterol turns a 12-year-old’s heart into that of a 60-year-old. However, the story is no longer one of inevitable tragedy.
The future looks even more promising with the development of Gene Therapy and CRISPR. Scientists are working on “one-and-done” fixes that could theoretically repair the broken genes in the liver. Instead of taking pills or undergoing weekly blood-cleaning procedures (Apheresis), children might one day have their “garbage collection system” fixed permanently.
As we look forward, we must ask ourselves: how does our understanding of this “ultra-rare” condition help us understand the health of every human heart? By studying the most extreme version of high cholesterol, we are learning how to protect everyone from the “unrelenting accumulation” of heart disease. In the race against the clock, science is finally starting to take the lead, ensuring that a 12-year-old’s heart can stay young, healthy, and strong for a lifetime.
DEEP DIVE
Clinical Trajectories and Pathophysiological Drivers of Early Mortality in Homozygous Familial Hypercholesterolemia
A Comprehensive Research Analysis
Evidence-Based Cardiovascular Research
Overview and Definitions
Homozygous familial hypercholesterolemia (HoFH) represents the most catastrophic and phenotypically extreme manifestation of inherited lipid disorders. It is an ultra-rare genetic condition characterized by the near-complete inability of the liver to clear low-density lipoprotein (LDL) particles from the systemic circulation, resulting in plasma cholesterol levels that are profoundly elevated from birth.[1]
Historically, HoFH was defined by a classic clinical triad: untreated LDL cholesterol (LDL-C) concentrations exceeding 500 mg/dL (13 mmol/L), the presence of pathognomonic cutaneous and tendon xanthomas appearing within the first decade of life, and evidence of heterozygous familial hypercholesterolemia (HeFH) in both biological parents.[1]
The scientific understanding of the disease has undergone a significant paradigm shift in the last decade. Genetic analysis has revealed a broader phenotypic spectrum than previously appreciated, leading clinical bodies such as the European Atherosclerosis Society (EAS) to refine diagnostic thresholds. The 2023 EAS consensAus statement recommends that HoFH be clinically suspected in any individual with untreated LDL-C levels above 400 mg/dL (~10 mmol/L), as many patients with genetically confirmed biallelic mutations present with levels below the traditional 500 mg/dL cutoff.[5] This is particularly relevant in pediatric populations, where LDL-C levels may be lower due to diet or physiological growth but still represent an extreme cardiovascular threat.[6]
The genetic architecture of HoFH is predominantly characterized by biallelic mutations in the LDLR gene, which encodes the LDL receptor responsible for hepatic uptake of cholesterol-rich particles.[8] These biallelic states may be true homozygous—involving the inheritance of the same pathogenic variant from both parents—or compound heterozygous, involving two distinct mutations in the same gene.[8] Beyond LDLR, rarer forms of HoFH are caused by pathogenic variants in the apolipoprotein B (APOB) gene, which impairs the ability of the LDL particle to bind to its receptor, and gain-of-function mutations in the proprotein convertase subtilisin/kexin type 9 (PCSK9) gene, which accelerates the intracellular degradation of the LDL receptor.[10] Additionally, an autosomal recessive form of the disease (ARH) results from biallelic loss-of-function variants in the LDLRAP1 gene.[8]
The prevalence of HoFH was traditionally estimated at 1 in 1,000,000 individuals, but contemporary epidemiological data and registry findings suggest a higher frequency of approximately 1 in 160,000 to 1 in 300,000.[1,6] In specific populations with founder effects—such as French Canadians, Lebanese Christians, South African Afrikaners, and certain Ashkenazi Jewish communities—the prevalence can be significantly higher.[16]
Differential diagnosis is critical. Sitosterolemia (phytosterolaemia), caused by biallelic variants in the ABCG5/ABCG8 genes, presents with extreme elevations in LDL-C and early-onset xanthomas but responds remarkably well to dietary modification and ezetimibe. Lysosomal acid lipase deficiency (LAL-D) also leads to severe hypercholesterolemia and premature atherosclerosis but is managed with enzyme replacement therapy. Cerebrotendinous xanthomatosis (CTX) may present with xanthomas similar to HoFH, though cholesterol levels are typically normal to mildly elevated.[1]
Mechanism of Extreme Risk in Early Life
The extreme cardiovascular risk in HoFH is driven by the unrelenting accumulation of cholesterol within the arterial wall, starting in utero. The central concept in quantifying this risk is the “cumulative cholesterol burden,” defined as the total mass of LDL-C to which the arterial intima has been exposed over time.[19]
The cumulative burden is expressed in mmol/L-years. Clinical evidence indicates that the onset of clinical atherosclerotic cardiovascular disease (ASCVD) in the general population corresponds to a cumulative threshold of approximately 125 mmol/L-years (~4,800 mg/dL-years).[62,63], while the threshold for first myocardial infarction is approximately 160 mmol/L-years (~6,000 mg/dL-years). A normolipidemic individual typically reaches the 160 mmol/L-year burden around age 55. In contrast, an untreated HoFH patient with an LDL-C of ~500 mg/dL (~13 mmol/L) reaches this same threshold by approximately age 12, explaining why children with the most severe null-receptor mutations often experience myocardial infarctions or sudden cardiac death before the age of 10.[19,63]
The Vascular Pathology of Pediatric HoFH
The pathophysiology of atherosclerosis in pediatric HoFH involves rapid progression from endothelial activation to advanced plaque formation. Under the influence of extreme hypercholesterolemia, LDL particles penetrate the arterial intima, where they are oxidized and engulfed by macrophages to form foam cells.[24] In the general population, this process takes decades to progress from fatty streaks to obstructive plaques. In HoFH, the sheer flux of lipoproteins into the arterial wall is so high that these fatty streaks evolve into complex, lipid-rich, and highly inflammatory plaques within a few years of life.[19]
A distinguishing feature of HoFH-associated vascular disease is its anatomical predilection for the aortic root and the coronary ostia.[27] The cholesterol-rich nature of these lesions frequently results in supravalvular aortic stenosis (SVAS). Unlike the degenerative valvular calcification seen in elderly populations, SVAS in HoFH is characterized by massive infiltration of the aortic wall and valve leaflets by xanthomatous tissue.[20,21] Furthermore, plaque deposition at the coronary ostia can lead to sudden, total occlusion even in children who do not yet have diffuse coronary artery disease.[27]
The Role of Secondary Risk Modifiers
In addition to the primary LDL-C elevation, many HoFH patients have significantly elevated levels of Lipoprotein(a) [Lp(a)]. Lp(a) is an LDL-like particle with an added apolipoprotein(a) moiety, conferring pro-thrombotic and pro-inflammatory properties. Because Lp(a) is cleared primarily via the LDL receptor, its levels are frequently twice as high in HoFH patients compared to the general population.[2] The presence of high Lp(a) acts as a risk multiplier, further accelerating atherosclerosis and increasing the likelihood of plaque rupture or thrombosis at a young age.[11]
The development of cutaneous xanthomas before the age of 4 is associated with a markedly increased risk of childhood coronary heart disease and early mortality.[20] These deposits are not merely cosmetic; they represent the systemic spillover of cholesterol that the vascular system can no longer contain.[2]
Magnitude of Lipid Abnormalities
The magnitude of hypercholesterolemia in HoFH is unparalleled in clinical medicine. Untreated total cholesterol levels typically range from 460 to 1,160 mg/dL (12–30 mmol/L), with LDL-C levels consistently exceeding 400 mg/dL (10 mmol/L) in most genetically confirmed cases.[4,5] In extreme cases, particularly in individuals with null-receptor mutations, LDL-C can reach levels above 1,000 mg/dL (26 mmol/L), and the blood may take on a visually distinct, opaque appearance due to the extreme concentration of lipoproteins.[2]
Apolipoprotein B and Particle Metrics
Apolipoprotein B (ApoB) is the primary structural protein of all atherogenic lipoproteins, including VLDL, IDL, Lp(a), and LDL. Because there is exactly one ApoB molecule per particle, its measurement provides a direct count of the total number of atherogenic particles in the circulation.[35] The normal reference range for ApoB in adults is below 130 mg/dL, with a US population median of approximately 93 mg/dL. In HoFH, ApoB levels are typically four to six times higher than this reference range.[35]
The pathophysiology of ApoB in HoFH is defined by two factors: overproduction and severely impaired clearance. The absence of functional LDL receptors leads to a prolonged residence time for LDL particles in the blood—approximately 5–6 days compared to the normal 2.5 days.[70] This extended circulation time results in the particles becoming increasingly modified. While Pattern B (small, dense LDL) is generally more atherogenic because it more easily penetrates the arterial wall, in HoFH, the overwhelming mass of even larger LDL particles drives constant flux into the sub-endothelial space.[39]
Lipid Profile Comparison by Genotype
The severity of the lipid abnormality is intrinsically linked to the functional status of the LDL receptor. Patients are categorized as receptor-negative (less than 2% of normal LDLR activity) or receptor-defective (2% to 25% of normal activity).[1,56]
| Parameter | Receptor-Negative HoFH | Receptor-Defective HoFH | Reference Range |
| Untreated LDL-C | >600 mg/dL (>15.5 mmol/L) | 300–500 mg/dL (7.8–12.9 mmol/L) | <130 mg/dL (<3.4 mmol/L) |
| Untreated ApoB | >400 mg/dL (~4–6× normal) | 200–400 mg/dL (~2–4× normal) | <130 mg/dL |
| Untreated Lp(a) | Frequently markedly elevated | Elevated | <30 mg/dL |
| LDLR Activity | <2% of normal | 2%–25% of normal | 100% (normal) |
| Response to Statins | Minimal (~14% LDL-C reduction) | Moderate (~23% LDL-C reduction) | High (40–55% reduction) |
Table 1. Lipid profile comparison by LDLR genotype. Source: [1,3,41,71]
A study of the CASCADE FH Registry in the United States demonstrated that untreated LDL-C levels were significantly higher in patients enrolled as children (median 776 mg/dL, IQR 704–892) compared to those enrolled as adults (median 533 mg/dL, IQR 467–702; p=0.001).[31] This discrepancy likely reflects a survival bias, where children with the most extreme elevations are diagnosed early because they develop visible symptoms—xanthomas or early cardiac events—while those with milder elevations may go undiagnosed until adulthood.[23]
The LDL-C:ApoB Ratio and Discordance
The LDL-C:ApoB ratio is a surrogate for LDL particle size and cholesterol content. A ratio below 1.2 (expressed as mg/dL:mg/dL) indicates a predominance of small, dense LDL particles, which are highly susceptible to oxidation and more readily trapped within arterial proteoglycans.[38] In true HoFH, however, LDL-C and ApoB are typically both concordantly high, meaning that the extreme risk is driven primarily by the sheer volume of cholesterol mass and particle number rather than a shift in particle size alone.[18]
Clinical Severity and Natural History (Untreated)
The natural history of untreated HoFH is characterized by rapid, progressive atherosclerosis and a starkly shortened lifespan. Without intervention, the average age of death has historically been reported as approximately 18 years, with some children succumbing to myocardial infarction as early as age 5.[17,31]
The Evolution of Clinical Manifestations
The first clinical signs are usually dermatological. Cutaneous xanthomas often appear in the first year of life, presenting as soft, yellow nodular lesions at sites of friction, such as the elbows, knees, and buttocks.[8] Tendon xanthomas involve the thickening of the Achilles tendon and the extensor tendons of the hands.[8]
As the cholesterol burden increases, vascular and valvular manifestations emerge. Arcus lipoides corneae and xanthelasmas are frequently observed before age 10.[15] By the second decade, most untreated patients have developed symptomatic coronary artery disease and/or aortic root disease.[2]
Historical Case Evidence
In 1889, G. Lehzen and K. Knauss described two sisters who are now recognized as likely the first documented cases of HoFH.[28] The 11-year-old sister developed multiple yellow spots and lumps beginning at age 3. By age 11, she presented with “egg-like” tumors on her hands and massive xanthomas on her Achilles tendons. Clinical examination revealed a long blowing systolic murmur, and she died shortly after a surgery.[28]
The autopsy findings documented:
- Aorta: Thickened with fat-containing tissue and sclerotic plaques.
- Aortic Valve: Stenotic due to massive intima changes.
- Carotid Artery: The left carotid was nearly completely occluded.
- Coronary Arteries: Both showed multiple plaques, with the left being severely affected.[28]
Registry-Based Observations on Natural History
Data from contemporary registries in non-high-income countries, where access to advanced therapies is limited, reflect this historical pattern. In a study of 751 HoFH patients across 38 countries, those in non-high-income regions experienced their first major adverse cardiovascular event a decade earlier than those in high-income countries (median age 24 vs. 35 years).[47]
| Clinical Feature | Typical Age of Onset (Untreated) | Pathological Significance |
| Cutaneous Xanthomas | <1 year | Marker of extreme systemic saturation |
| Corneal Arcus | <10 years | Early indicator of lipid spillover |
| Tendon Xanthomas | 5–15 years | Cumulative tissue deposition |
| Aortic Root Stenosis | 5–20 years | Primary cause of non-ischemic cardiac death |
| Myocardial Infarction | 5–30 years | Result of ostial or diffuse CAD |
| Sudden Cardiac Death | Variable; can occur in childhood | Fatal arrhythmia or total ostial occlusion |
Table 2. Clinical progression of untreated HoFH. Source: [2,19,47]
Why Some Patients Survive Longer Than Others
Despite the severe nature of the disease, there is significant inter-individual variability in survival. Some patients succumb to cardiac arrest in early childhood, while others, even with identical genetic mutations, may live into their 50s or 60s. This heterogeneity is driven by a complex interplay of residual receptor activity, genetic modifiers, and the timing of therapeutic intervention.[1]
Residual LDLR Activity and Mutation Type
The most powerful predictor of clinical outcome is the residual activity of the LDL receptor. Receptor-negative individuals (null/null; <2% activity) exhibit the highest LDL-C levels, the poorest response to traditional medications, and the earliest onset of ASCVD.[3,56] Receptor-defective patients (2%–25% activity) often have LDL-C levels approximately 18% lower than receptor-negative patients and respond more vigorously to pharmacological up-regulation of the receptor.[41] This genetic dosing effect can delay the onset of cardiovascular events by a decade or more.[6]
Protective Genetic Modifiers
PCSK9 Loss-of-Function (LOF)
PCSK9 is a protein that binds to the LDL receptor and targets it for lysosomal degradation. Individuals who co-inherit a loss-of-function variant in PCSK9 have naturally lower circulating PCSK9 levels, leading to a higher density of LDL receptors on the hepatocyte surface.[13] Population studies show that PCSK9 LOF variants are associated with approximately 28% lower LDL-C and up to 88% reduction in CHD risk.[69] In the context of HoFH, a PCSK9 LOF variant can significantly counteract the effect of a pathogenic LDLR variant, resulting in a much milder clinical phenotype than would otherwise be expected.[14]
ANGPTL3 Loss-of-Function
Angiopoietin-like protein 3 (ANGPTL3) inhibits lipoprotein lipase and endothelial lipase. LOF variants in ANGPTL3 lead to lower levels of LDL, VLDL, and HDL through mechanisms largely independent of the LDL receptor.[9] Because this pathway does not rely on LDLR, it acts as a potent modifier even in receptor-negative HoFH patients.[9]
APOB Truncations
Some patients carry hypobetalipoproteinemia variants in the APOB gene that lead to reduced production of LDL particles. If a patient with an LDLR mutation also carries one of these variants, the liver produces fewer atherogenic vehicles, limiting the maximum LDL-C level achievable.[54]
Therapeutic Era and Intervention Thresholds
Before the 1980s, treatment was limited to low-fat diets and early bile acid sequestrants, which were largely ineffective.[17] The introduction of statins, and later LDL apheresis, began to extend the life expectancy of HoFH patients into the late 20s and 30s.[17]
The modern era, characterized by the availability of receptor-independent therapies, has fundamentally changed the prognosis. Lomitapide (an MTP inhibitor that reduces VLDL/LDL production) reduces LDL-C by approximately 50% independent of receptor genotype.[55,72] Evinacumab (an ANGPTL3 inhibitor) reduces LDL-C by 43–53% even in null-receptor patients.[32] The combination of these agents, if started in early childhood, allows many patients to keep their cumulative cholesterol burden below critical thresholds for much longer.[19]
Longest-Lived HoFH Cases
While the historic life expectancy was under 20 years, contemporary literature now documents survivors living into their 50s, 60s, and beyond.
The 57-Year-Old Tokyo Case (Komuro et al., 1987)
One of the first longest-lived cases reported was a 57-year-old Japanese male described in 1987.[64] This patient was homozygous for an internalization defect in the LDL receptor (a Class 4 mutation), meaning the receptors could bind LDL but could not pull it into the cell. His LDL-C was 461 mg/dL—lower than typical null-receptor patients—which allowed him to survive into his late 50s despite the absence of modern statins during much of his life.[64]
The 59-Year-Old Czech Case (Novák et al., 2025)
A 2025 study reported on several atypical Czech HoFH patients, including a 59-year-old male compound heterozygote for the p.Phe114Ile and p.Gly592Glu LDLR variants.[49] This patient was not diagnosed with HoFH until age 41. Although he eventually required a coronary artery bypass graft at age 57 and had significant carotid narrowing, his survival was exceptional given his genetic makeup. His specific compound heterozygous combination evidently conferred enough residual LDLR activity to prevent childhood mortality despite decades of extreme LDL-C elevation.[49]
The 72-Year-Old Heterozygous FH Case (Johnson et al., 2018)
An instructive case involves a 72-year-old male with a pathogenic LDLR variant (p.Val827Ile) who maintained untreated LDL-C consistently around 487 mg/dL throughout his life.[37] Despite this extreme elevation and lifelong lack of treatment, he had an Agatston calcium score of 0 on multiple scans—implying a complete absence of coronary artery calcification. His survival was attributed to an exceptionally high HDL-C (~68 mg/dL) and a Pattern A LDL (large, buoyant particles), which are less prone to oxidation and arterial retention.[37] Editorial note: This patient has been confirmed as heterozygous FH, not HoFH, based on the primary publication.[37] The case is cited here as an extreme FH phenotype illustrating the protective role of HDL-C and LDL particle size.
Contemporary Pediatric Successes
Registry data now show that children diagnosed at age 2 and started on aggressive therapy—including liver transplantation or apheresis—are reaching adulthood with minimal atherosclerotic burden.[23] Case reports document patients managed with weekly plasmapheresis and LDL apheresis from their teens into their 30s, maintaining a high quality of life and illustrating that mechanical clearance of cholesterol can effectively substitute for missing hepatic receptors.[59]
Summary Diagnostic and Clinical Tables
Table 3: Refined Clinical and Genetic Criteria for HoFH Diagnosis
| Category | Diagnostic Threshold / Feature | Rationale |
| Untreated LDL-C | >400 mg/dL (>10 mmol/L) | Proposed by 2023 EAS to capture broader spectrum |
| Treated LDL-C | >300 mg/dL (>8 mmol/L) on statin + ezetimibe | Historical threshold; used when baseline is unknown |
| Genetic Criteria | Bi-allelic variants (LDLR, APOB, PCSK9, LDLRAP1) | Gold standard for confirmation |
| Physical Findings | Xanthomas before age 10; Corneal Arcus | Pathognomonic for extreme cumulative burden |
| Family History | HeFH in both biological parents | Consistent with autosomal codominant pattern |
| Non-Genetic Mimics | Sitosterolaemia; LAL-D; CTX | Must be excluded to ensure appropriate therapy |
Source: [1,5]
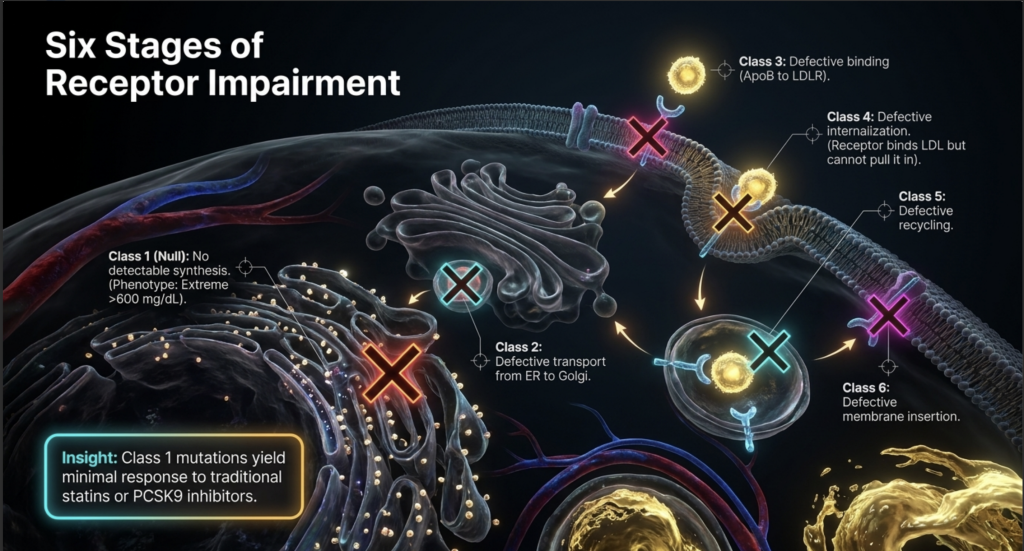
Table 4: Functional Classification of LDLR Mutations in HoFH
| Class | Mechanism of Defect | Phenotypic Severity | Therapeutic Implication |
| Class 1 | No detectable synthesis (Null) | Extreme (>600 mg/dL) | No response to Statins/PCSK9i |
| Class 2 | Defective transport (ER to Golgi) | Severe | Minimal drug response |
| Class 3 | Defective binding (ApoB to LDLR) | Variable | May respond to PCSK9i |
| Class 4 | Defective internalization | Moderate to Severe | Some residual clearance possible |
| Class 5 | Defective recycling | Moderate | Often responds to drug up-regulation |
| Class 6 | Defective membrane insertion | Variable | Depends on density of insertion |
Source: [3]
Table 5: Pediatric vs. Adult HoFH — CASCADE FH Registry (Cuchel et al., 2023)
| Metric | Children (n=16) Median (IQR) | Adults (n=51) Median (IQR) |
| Age at Diagnosis (years) | 2.5 (1–6) | 23 (13–34) |
| Untreated LDL-C (mg/dL) | 776 (704–892) | 533 (467–702) |
| ASCVD at Enrollment | 43.8% | 78.4% |
| Aortic Valve Stenosis at Enrollment | 18.8% | 25.5% |
| CABG (any) | 12.5% | 41.2% |
Source: [31]
Evidence Quality and Uncertainty
The scientific community’s understanding of HoFH has evolved from descriptive case studies to comprehensive international registries, but several critical areas of uncertainty remain.
Limitations of Current Evidence
The rarity of HoFH makes large-scale, randomized controlled trials (RCTs) extremely difficult. Much of the evidence for long-term survival and treatment efficacy is derived from retrospective registry data (such as CASCADE FH or the Worldwide HoFH Study) or open-label phase 2 and phase 3 trials.[17] While registries provide invaluable real-world data, they are subject to selection bias—patients who are more severely affected or who have access to specialized care are more likely to be enrolled.[1]
A significant area of uncertainty is the missing genetic cause in approximately 20–40% of patients with a clinical diagnosis of HoFH.[7,66] This suggests that there are either unidentified FH genes or that a polygenic mechanism—the accumulation of many small-effect variants—can mimic the severity of monogenic HoFH. The clinical management of these mutation-negative patients remains a challenge.[67,68]
Gaps in Pediatric Management
While current guidelines recommend universal lipid screening in children, the optimal age for initiating advanced therapies (such as evinacumab or lomitapide) in toddlers is still under investigation.[1] The long-term safety of these agents in developing children is a concern, yet the risk of waiting for more data is the development of irreversible aortic root disease.[27]
Furthermore, the threshold hypothesis of cumulative cholesterol burden (expressed in mmol/L-years) is an elegant model but has not been prospectively validated as a definitive point of no return.[19] There is debate over whether lowering LDL-C can actually regress existing pediatric plaques or merely prevent the formation of new ones.[25]
Future Directions
The future of HoFH research lies in gene therapy and base editing. Agents intended to directly alter the LDLR or PCSK9 genes in the liver—including in vivo CRISPR base editing—are currently in preclinical and early clinical trials.[52,53] These one-and-done therapies could theoretically eliminate the need for lifelong infusions and daily pills, but the long-term genomic stability and safety of these approaches remain the primary uncertainties of the coming decade.[52,53]
Conclusion
HoFH is a life-threatening emergency that manifests in the first years of life. The magnitude of the lipid abnormality is so great that it overcomes standard biological repair mechanisms, necessitating a multi-hit, receptor-independent therapeutic approach. Survival variability is driven by the specific nature of the genetic defect and the presence of protective modifiers, but the most important determinant of life or death remains the timing of diagnosis and the intensity of LDL-C lowering achieved in the first decade of life.[1,19,31]
References
- Cuchel M, Bruckert E, Ginsberg HN, et al. Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur Heart J. 2014;35(32):2146-2157. doi:10.1093/eurheartj/ehu274
- Rahman A, Ahmed MU, Islam AK, Karim A, Sarker SA. A young male with familial hypercholesterolemia. J Saudi Heart Assoc. 2012;24(4):261-264. doi:10.1016/j.jsha.2012.06.264
- Suryawanshi YN, Warbhe RA. Familial Hypercholesterolemia: A Literature Review of the Pathophysiology and Current and Novel Treatments. Cureus. 2023;15(11):e49121. Published 2023 Nov 20. doi:10.7759/cureus.49121
- Ziajka PE. Management of patients with homozygous familial hypercholesterolemia. Am J Manag Care. 2013;19(13 Suppl):.
- Cuchel M, Raal FJ, Hegele RA, et al. 2023 Update on European Atherosclerosis Society Consensus Statement on Homozygous Familial Hypercholesterolaemia: new treatments and clinical guidance. Eur Heart J. 2023;44(25):2277-2291. doi:10.1093/eurheartj/ehad197
- Sjouke B, Kusters DM, Kindt I, et al. Homozygous autosomal dominant hypercholesterolaemia in the Netherlands: prevalence, genotype-phenotype relationship, and clinical outcome. Eur Heart J. 2015;36(9):560-565. doi:10.1093/eurheartj/ehu058
- Sturm AC, Knowles JW, Gidding SS, et al. Clinical Genetic Testing for Familial Hypercholesterolemia: JACC Scientific Expert Panel. J Am Coll Cardiol. 2018;72(6):662-680. doi:10.1016/j.jacc.2018.05.044
- Marais AD. Familial hypercholesterolaemia. Clin Biochem Rev. 2004;25(1):49-68.
- van den Bosch SE, Corpeleijn WE, Hutten BA, Wiegman A. How Genetic Variants in Children with Familial Hypercholesterolemia Not Only Guide Detection, but Also Treatment. Genes (Basel). 2023;14(3):669. Published 2023 Mar 7. doi:10.3390/genes14030669
- Zhao L, Gao Y, Liu G, et al. Zhonghua Xin Xue Guan Bing Za Zhi. 2022;50(6):585-590. doi:10.3760/cma.j.cn112148-20210715-00591
- Warden BA, Fazio S, Shapiro MD. Familial Hypercholesterolemia: Genes and Beyond. In: Feingold KR, Adler RA, Ahmed SF, et al., eds. Endotext. South Dartmouth (MA): MDText.com, Inc.; September 23, 2024.
- Razman AZ, Chua YA, Mohd Kasim NA, et al. Genetic Spectrum of Familial Hypercholesterolaemia in the Malaysian Community: Identification of Pathogenic Gene Variants Using Targeted Next-Generation Sequencing. Int J Mol Sci. 2022;23(23):14971. Published 2022 Nov 29. doi:10.3390/ijms232314971
- Saavedra YG, Dufour R, Davignon J, Baass A. PCSK9 R46L, lower LDL, and cardiovascular disease risk in familial hypercholesterolemia: a cross-sectional cohort study. Arterioscler Thromb Vasc Biol. 2014;34(12):2700-2705. doi:10.1161/ATVBAHA.114.304406
- Bayona A, Arrieta F, Rodríguez-Jiménez C, et al. Loss-of-function mutation of PCSK9 as a protective factor in the clinical expression of familial hypercholesterolemia: A case report. Medicine (Baltimore). 2020;99(34):e21754. doi:10.1097/MD.0000000000021754
- Gidding SS, Champagne MA, de Ferranti SD, et al. The Agenda for Familial Hypercholesterolemia: A Scientific Statement From the American Heart Association. Circulation. 2015;132(22):2167-2192. doi:10.1161/CIR.0000000000000297
- Defesche JC, Gidding SS, Harada-Shiba M, Hegele RA, Santos RD, Wierzbicki AS. Familial hypercholesterolaemia. Nat Rev Dis Primers. 2017;3:17093. Published 2017 Dec 7. doi:10.1038/nrdp.2017.93
- Raal FJ, Pilcher GJ, Panz VR, et al. Reduction in mortality in subjects with homozygous familial hypercholesterolemia associated with advances in lipid-lowering therapy. Circulation. 2011;124(20):2202-2207. doi:10.1161/CIRCULATIONAHA.111.042523
- Goldberg AC, Hopkins PN, Toth PP, et al. Familial hypercholesterolemia: screening, diagnosis and management of pediatric and adult patients: clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. 2011;5(3 Suppl):S1-S8. doi:10.1016/j.jacl.2011.04.003
- Wiegman A, Gidding SS, Watts GF, et al. Familial hypercholesterolaemia in children and adolescents: gaining decades of life by optimizing detection and treatment. Eur Heart J. 2015;36(36):2425-2437. doi:10.1093/eurheartj/ehv157
- Bélanger AM, Akioyamen LE, Ruel I, Hales L, Genest J. Aortic stenosis in homozygous familial hypercholesterolaemia: a paradigm shift over a century. Eur Heart J. 2022;43(34):3227-3239. doi:10.1093/eurheartj/ehac339
- Rocha VZ, Santos RD. Past, Present, and Future of Familial Hypercholesterolemia Management. Methodist Debakey Cardiovasc J. 2021;17(4):28-35. Published 2021 Sep 24. doi:10.14797/mdcvj.887
- Cuchel M, Raal FJ, Hegele RA, et al. 2023 Update on European Atherosclerosis Society Consensus Statement on Homozygous Familial Hypercholesterolaemia: new treatments and clinical guidance. Eur Heart J. 2023;44(25):2277-2291. doi:10.1093/eurheartj/ehad197
- Family Heart Foundation, “Family Heart Foundation study shows some children with HoFH miss out on decades of life-saving treatment,” EurekAlert!, 2022. [Press release; cited for registry context only.]
- Hong YM. Atherosclerotic cardiovascular disease beginning in childhood. Korean Circ J. 2010;40(1):1-9. doi:10.4070/kcj.2010.40.1.1
- Tousoulis D, Kampoli AM, Papageorgiou N, et al. Pathophysiology of atherosclerosis: the role of inflammation. Curr Pharm Des. 2011;17(37):4089-4110. doi:10.2174/138161211798764843
- Wiegman A, Gidding SS, Watts GF, et al. Familial hypercholesterolaemia in children and adolescents: gaining decades of life by optimizing detection and treatment. Eur Heart J. 2015;36(36):2425-2437. doi:10.1093/eurheartj/ehv157
- Cuchel M, Raal FJ, Hegele RA, et al. 2023 Update on European Atherosclerosis Society Consensus Statement on Homozygous Familial Hypercholesterolaemia: new treatments and clinical guidance. Eur Heart J. 2023;44(25):2277-2291. doi:10.1093/eurheartj/ehad197
- Ballantyne CM, Gellis L, Tardif JC, et al. Efficacy and Safety of Oral PCSK9 Inhibitor Enlicitide in Adults With Heterozygous Familial Hypercholesterolemia: A Randomized Clinical Trial. JAMA. 2026;335(2):129-139. doi:10.1001/jama.2025.20620
- Maldar SB, Pinto CJ. Homozygous familial hypercholesterolaemia in a patient presenting with hypertensive encephalopathy. BMJ Case Rep. 2022;15(10):e250265. Published 2022 Oct 31. doi:10.1136/bcr-2022-250265
- Suresh Kumar G, Mathbout MF, Fahsah I, Ghafghazi S. Case of homozygous familial hypercholesterolaemia with premature coronary artery disease. BMJ Case Rep. 2021;14(5):e242114. Published 2021 May 19. doi:10.1136/bcr-2021-242114
- Cuchel M, Lee PC, Hudgins LC, et al. Contemporary Homozygous Familial Hypercholesterolemia in the United States: Insights From the CASCADE FH Registry. J Am Heart Assoc. 2023;12(9):e029175. doi:10.1161/JAHA.122.029175
- Raal FJ, Rosenson RS, Reeskamp LF, et al. Evinacumab for Homozygous Familial Hypercholesterolemia. N Engl J Med. 2020;383(8):711-720. doi:10.1056/NEJMoa2004215
- Palacio CH, Harring TR, Nguyen NT, Goss JA, O’Mahony CA. Homozygous familial hypercholesterolemia: case series and review of the literature. Case Rep Transplant. 2011;2011:154908. doi:10.1155/2011/154908
- Raal FJ, Santos RD. Homozygous familial hypercholesterolemia: current perspectives on diagnosis and treatment. Atherosclerosis. 2012;223(2):262-268. doi:10.1016/j.atherosclerosis.2012.02.019
- Contois JH, McConnell JP, Sethi AA, et al. Apolipoprotein B and cardiovascular disease risk: position statement from the AACC Lipoproteins and Vascular Diseases Division Working Group on Best Practices. Clin Chem. 2009;55(3):407-419. doi:10.1373/clinchem.2008.118356
- Cuchel M, Bruckert E, Ginsberg HN, et al. Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur Heart J. 2014;35(32):2146-2157. doi:10.1093/eurheartj/ehu274
- Johnson KW, Dudley JT, Bobe JR. A 72-Year-Old Patient with Longstanding, Untreated Familial Hypercholesterolemia but no Coronary Artery Calcification: A Case Report. Cureus. 2018;10(4):e2452. Published 2018 Apr 9. doi:10.7759/cureus.2452
- Nordestgaard BG, Langsted A, Mora S, et al. Fasting is not routinely required for determination of a lipid profile: clinical and laboratory implications including flagging at desirable concentration cut-points-a joint consensus statement from the European Atherosclerosis Society and European Federation of Clinical Chemistry and Laboratory Medicine. Eur Heart J. 2016;37(25):1944-1958. doi:10.1093/eurheartj/ehw152
- Pencina KM, Pencina MJ, Lawler PR, et al. Interplay of Atherogenic Particle Number and Particle Size and the Risk of Coronary Heart Disease. Clin Chem. 2023;69(1):48-55. doi:10.1093/clinchem/hvac172
- Drouin-Chartier JP, Tremblay AJ, Bergeron J, Lamarche B, Couture P. The Low-Density Lipoprotein Receptor Genotype Is a Significant Determinant of the Rebound in Low-Density Lipoprotein Cholesterol Concentration After Lipoprotein Apheresis Among Patients With Homozygous Familial Hypercholesterolemia. Circulation. 2017;136(9):880-882. doi:10.1161/CIRCULATIONAHA.117.029435
- Bertolini S, Cantafora A, Averna M, et al. Clinical expression of familial hypercholesterolemia in clusters of mutations of the LDL receptor gene that cause a receptor-defective or receptor-negative phenotype. Arterioscler Thromb Vasc Biol. 2000;20(9):E41-E52. doi:10.1161/01.atv.20.9.e41
- Jayaram S, Meera S, Kadi S, Sreenivasa N. An Interesting Case of Familial Homozygous Hypercholesterolemia-A Brief Review. Indian J Clin Biochem. 2012;27(3):309-313. doi:10.1007/s12291-011-0165-8
- Bensabbahia D, El Achiwi M, Atrassi M, Abkari A, Widad G. Homozygous Familial Hypercholesterolemia in a Seven-Year-Old: A Case Study Highlighting the Importance of Early Diagnosis. Cureus. 2025;17(6):e86219. Published 2025 Jun 17. doi:10.7759/cureus.86219
- Mainieri F, Tagi VM, Chiarelli F. Recent Advances on Familial Hypercholesterolemia in Children and Adolescents. Biomedicines. 2022;10(5):1043. Published 2022 Apr 30. doi:10.3390/biomedicines10051043
- Grundy SM, Stone NJ; Guideline Writing Committee for the 2018 Cholesterol Guidelines. 2018 Cholesterol Clinical Practice Guidelines: Synopsis of the 2018 American Heart Association/American College of Cardiology/Multisociety Cholesterol Guideline. Ann Intern Med. 2019;170(11):779-783. doi:10.7326/M19-0365
- Defesche JC, Gidding SS, Harada-Shiba M, Hegele RA, Santos RD, Wierzbicki AS. Familial hypercholesterolaemia. Nat Rev Dis Primers. 2017;3:17093. Published 2017 Dec 7. doi:10.1038/nrdp.2017.93
- Tromp TR, Hartgers ML, Hovingh GK, et al. Worldwide experience of homozygous familial hypercholesterolaemia: retrospective cohort study. Lancet. 2022;399(10326):719-728. doi:10.1016/S0140-6736(21)02001-8
- Vladimirova-Kitova L, Kitov S, Ganev M, Chochkova-Bukova L. Case Report: Difficulties in the Treatment of a 12-Year-Old Patient With Homozygous Familial Hypercholesterolemia, Compound Heterozygous Form – 5 Years Follow-Up. Front Cardiovasc Med. 2021;8:743341. Published 2021 Oct 8. doi:10.3389/fcvm.2021.743341
- Zlatohlávek L, Becherová Beňová J, Foglarová T, Dudková T, Hubáček JA. Case Report: Beating the assumed prognosis: homozygous familial hypercholesterolemia with unexpected long survival. Front Cardiovasc Med. 2025;12:1643771. Published 2025 Oct 20. doi:10.3389/fcvm.2025.1643771
- Guo Q, Feng X, Zhou Y. PCSK9 Variants in Familial Hypercholesterolemia: A Comprehensive Synopsis. Front Genet. 2020;11:1020. Published 2020 Sep 23. doi:10.3389/fgene.2020.01020
- Bayona A, Arrieta F, Rodríguez-Jiménez C, et al. Loss-of-function mutation of PCSK9 as a protective factor in the clinical expression of familial hypercholesterolemia: A case report. Medicine (Baltimore). 2020;99(34):e21754. doi:10.1097/MD.0000000000021754
- Musunuru K, Chadwick AC, Mizoguchi T, et al. In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature. 2021;593(7859):429-434. doi:10.1038/s41586-021-03534-y
- Lee RG, Mazzola AM, Braun MC, et al. Efficacy and Safety of an Investigational Single-Course CRISPR Base-Editing Therapy Targeting PCSK9 in Nonhuman Primate and Mouse Models. Circulation. 2023;147(3):242-253. doi:10.1161/CIRCULATIONAHA.122.062132
- Lacaze P, Riaz M, Sebra R, et al. Protective lipid-lowering variants in healthy older individuals without coronary heart disease. Open Heart. 2021;8(2):e001710. doi:10.1136/openhrt-2021-001710
- Cuchel M, Meagher EA, du Toit Theron H, et al. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: a single-arm, open-label, phase 3 study. Lancet. 2013;381(9860):40-46. doi:10.1016/S0140-6736(12)61731-0
- Norata GD, Tibolla G, Catapano AL. PCSK9 inhibition for the treatment of hypercholesterolemia: promises and emerging challenges. Vascul Pharmacol. 2014;62(2):103-111. doi:10.1016/j.vph.2014.05.011
- Bytyçi I, Henein MY, Bytyqi S, et al. PCSK9 and ANGPTL3 Inhibitors in Homozygous Familial Hypercholesterolemia: A Meta-analysis of Randomized Clinical Trials. Drugs. 2026;86(2):231-242. doi:10.1007/s40265-025-02272-z
- Banerjee P, Chan KC, Tarabocchia M, et al. Functional Analysis of LDLR (Low-Density Lipoprotein Receptor) Variants in Patient Lymphocytes to Assess the Effect of Evinacumab in Homozygous Familial Hypercholesterolemia Patients With a Spectrum of LDLR Activity. Arterioscler Thromb Vasc Biol. 2019;39(11):2248-2260. doi:10.1161/ATVBAHA.119.313051
- Alicezah MK, Razali R, Rahman T, et al. Homozygous familial hypercholesterolemia. Malays J Pathol. 2014;36(2):131-137.
- Harada-Shiba M, Ohtake A, Sugiyama D, et al. Guidelines for the Diagnosis and Treatment of Pediatric Familial Hypercholesterolemia 2022. J Atheroscler Thromb. 2023;30(5):531-557. doi:10.5551/jat.CR006
- Bélanger AM, Akioyamen L, Alothman L, Genest J. Evidence for improved survival with treatment of homozygous familial hypercholesterolemia. Curr Opin Lipidol. 2020;31(4):176-181. doi:10.1097/MOL.0000000000000686
- Ference BA, Graham I, Tokgozoglu L, Catapano AL. Impact of Lipids on Cardiovascular Health: JACC Health Promotion Series. J Am Coll Cardiol. 2018;72(10):1141-1156. doi:10.1016/j.jacc.2018.06.046
- Nordestgaard BG, Chapman MJ, Humphries SE, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013;34(45):3478-90a. doi:10.1093/eurheartj/eht273
- Komuro I, Kato H, Nakagawa T, et al. The longest-lived patient with homozygous familial hypercholesterolemia secondary to a defect in internalization of the LDL receptor. Am J Med Sci. 1987;294(5):341-345. doi:10.1097/00000441-198711000-00008
- Beheshti SO, Madsen CM, Varbo A, Nordestgaard BG. Worldwide Prevalence of Familial Hypercholesterolemia: Meta-Analyses of 11 Million Subjects. J Am Coll Cardiol. 2020;75(20):2553-2566. doi:10.1016/j.jacc.2020.03.057
- Futema M, Taylor-Beadling A, Williams M, Humphries SE. Genetic testing for familial hypercholesterolemia-past, present, and future. J Lipid Res. 2021;62:100139. doi:10.1016/j.jlr.2021.100139
- Vrablik M, Tichý L, Freiberger T, Blaha V, Satny M, Hubacek JA. Genetics of Familial Hypercholesterolemia: New Insights. Front Genet. 2020;11:574474. Published 2020 Oct 7. doi:10.3389/fgene.2020.574474
- Di Taranto MD, Giacobbe C, Fortunato G. Familial hypercholesterolemia: A complex genetic disease with variable phenotypes. Eur J Med Genet. 2020;63(4):103831. doi:10.1016/j.ejmg.2019.103831
- Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354(12):1264-1272. doi:10.1056/NEJMoa054013
- Bilheimer DW, Stone NJ, Grundy SM. Metabolic studies in familial hypercholesterolemia. Evidence for a gene-dosage effect in vivo. J Clin Invest. 1979;64(2):524-533. doi:10.1172/JCI109490
- Stein EA, Gipe D, Bergeron J, et al. Effect of a monoclonal antibody to PCSK9, REGN727/SAR236553, to reduce low-density lipoprotein cholesterol in patients with heterozygous familial hypercholesterolaemia on stable statin dose with or without ezetimibe therapy: a phase 2 randomised controlled trial. Lancet. 2012;380(9836):29-36. doi:10.1016/S0140-6736(12)60771-5
- Blom DJ, Averna MR, Meagher EA, et al. Long-Term Efficacy and Safety of the Microsomal Triglyceride Transfer Protein Inhibitor Lomitapide in Patients With Homozygous Familial Hypercholesterolemia. Circulation. 2017;136(3):332-335. doi:10.1161/CIRCULATIONAHA.117.028208