
The Atherogenic Lipoprotein Hypothesis: A Comprehensive Review of Causal Evidence, Population Anomalies, Industry Interference, and the Organized Denial of Cholesterol Science
The historical evolution of cardiovascular epidemiology has been defined by the tension between observed population patterns and the underlying physiological mechanisms of disease. Atherosclerotic cardiovascular disease (ASCVD) remains the leading cause of global mortality, yet the “lipid hypothesis” — the proposition that circulating lipoproteins are the primary causal drivers of this pathology — remains a subject of intense heterodox challenge. This report provides a scientifically rigorous analysis of the evidence supporting the causality of apolipoprotein B-containing lipoproteins, reconciles historical and modern population anomalies within the lipid model, evaluates the credibility of industry-funded efforts to maintain scientific uncertainty, examines the organized commercial ecosystem that profits from cholesterol denialism, and documents the specific methodological errors deployed by groups motivated to disprove the importance of cholesterol in cardiovascular disease.
Section 1 — The ApoB vs. LDL-C Distinction: From Cholesterol Mass to Particle Burden
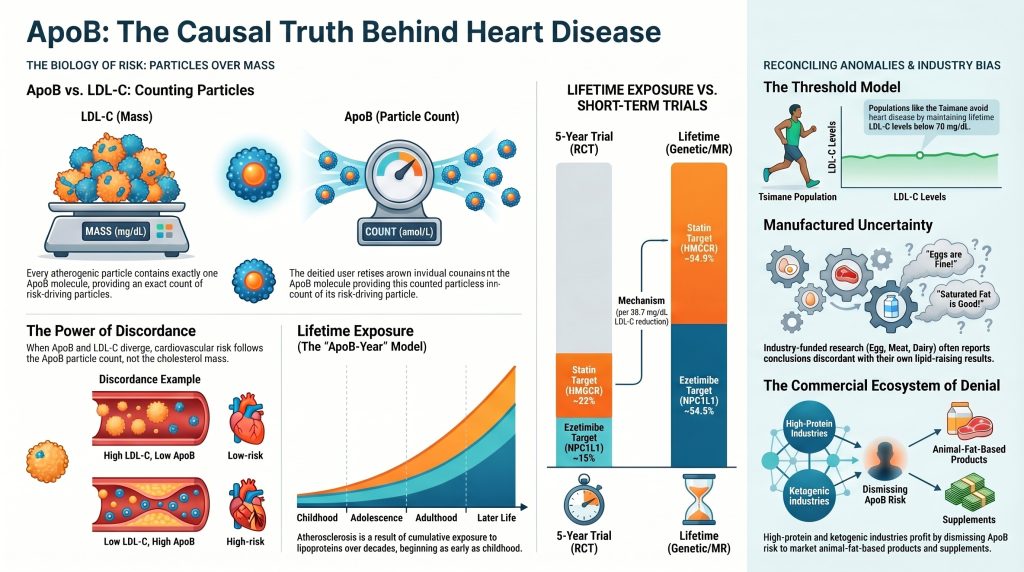
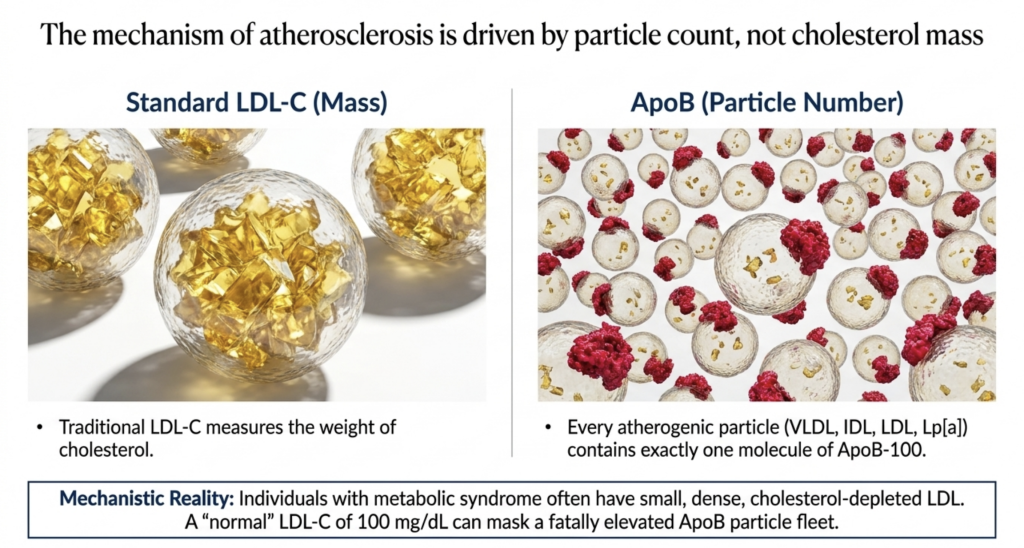
The traditional clinical reliance on low-density lipoprotein cholesterol (LDL-C) as the primary biomarker for cardiovascular risk is a historical legacy of the late 20th century. LDL-C measures the total mass of cholesterol contained within the low-density lipoprotein fraction, typically expressed in milligrams per deciliter (mg/dL) or millimoles per liter (mmol/L). While highly correlated with risk in most populations, LDL-C is fundamentally an imperfect proxy because it provides no direct information regarding the actual number of circulating atherogenic particles. Atherosclerosis is a disease of particle infiltration and entrapment; it is the physical number of lipoproteins entering the arterial wall, rather than the amount of cargo they carry, that dictates the rate of plaque progression.
Apolipoprotein B-100 (ApoB) is the structural protein found on the surface of every potentially atherogenic lipoprotein. Each particle — whether very-low-density lipoprotein (VLDL), intermediate-density lipoprotein (IDL), low-density lipoprotein (LDL), or lipoprotein(a) [Lp(a)] — contains exactly one molecule of ApoB.[1] Consequently, measuring ApoB provides an exact count of the total number of atherogenic particles in the plasma. The distinction between cholesterol mass (LDL-C) and particle burden (ApoB) becomes clinically critical in cases of “discordance,” where these two metrics diverge.
The Mechanism of Discordance and Particle Variability
Discordance typically arises due to variations in the cholesterol content of individual particles. In individuals with metabolic syndrome, insulin resistance, or hypertriglyceridemia, the intravascular remodeling of lipoproteins leads to the formation of small, dense LDL particles that are relatively depleted of cholesterol. In these individuals, a “normal” LDL-C level of 100 mg/dL may mask a significantly elevated ApoB count, as more particles are required to transport the same mass of cholesterol. Conversely, individuals with large, cholesterol-rich LDL particles may have a high LDL-C but a relatively lower ApoB, representing a lower total particle burden.
Nuclear Magnetic Resonance (NMR) spectroscopy has further refined this understanding by allowing for the direct measurement of LDL particle number (LDL-P) and size. Data from large prospective cohorts, including the AMORIS study, the EPIC-Norfolk cohort, the UK Biobank, and the Women’s Health Study, have consistently demonstrated that when ApoB and LDL-C are discordant, the cardiovascular risk tracks with the ApoB count (the particle number) rather than the LDL-C mass.[1] For example, individuals in the discordantly high ApoB group (low LDL-C, high ApoB) exhibit a significantly increased risk of major adverse cardiovascular events (MACE) compared to those with concordant levels.[4]
Table 1: LDL-C/ApoB Discordance and Risk of MACE — UK Biobank (n = 375,544)
| Discordance Category | Risk of MACE (Hazard Ratio) | Mediation by VLDL/Triglycerides | Clinical Interpretation |
| Concordant (LDL-C & ApoB) | 1.0 (Referent) | N/A | Standard Risk Assessment |
| Discordantly High ApoB | 1.11 [1.06–1.15] | 25.5% (VLDL), 26.6% (TG) | Risk Underestimated by LDL-C |
| Discordantly Low ApoB | 0.87 [0.83–0.93] | N/A | Risk Overestimated by LDL-C |
Source: UK Biobank NMR sub-study.[4]
In the UK Biobank analysis, the increased risk in individuals with discordantly high ApoB was partially mediated by the presence of VLDL particles and elevated triglycerides, which are often overlooked when focusing solely on LDL-C.[4] These findings reinforce the utility of ApoB as a more comprehensive marker of risk, particularly in primary prevention where traditional lipid panels may fail to identify high-risk individuals with metabolic dysfunction.
Section 2 — Populations Used to Challenge the Lipid Hypothesis: Reconciling Anomalies
Heterodox critiques of the lipid hypothesis often rely on “anomalous” populations that appear to have high fat intake or high cholesterol levels without a corresponding burden of cardiovascular disease. However, a mechanistic review using primary literature and autopsy data demonstrates that these anomalies are either artifacts of poor data quality or, in many cases, provide strong supporting evidence for the lipid hypothesis when evaluated correctly.
1. Inuit and Arctic Populations: The Myth of Immunity
The “Inuit Paradox” was popularized by Bang and Dyerberg, who suggested that Greenland Eskimos had exceptionally low rates of ischemic heart disease (IHD) despite a high-fat diet rich in marine mammals.[7] This was attributed to the high intake of omega-3 polyunsaturated fatty acids, which were hypothesized to provide an antithrombotic shield.[9]
However, the scientific evidence for low IHD among the Inuit is remarkably fragile and rests on unreliable mortality statistics. Reassessments of Greenlandic and Alaskan health data have highlighted several critical distortions. First, early mortality records in Greenland suffered from significant misclassification; a high proportion of cardiovascular deaths were attributed to “garbage codes” such as heart failure or ill-defined heart disease rather than IHD. When standardized algorithms are applied to these historical data, the difference in IHD mortality between Greenland and Denmark is markedly reduced.
Autopsy data provide the most definitive rebuttal. Studies from the 1960s and 1970s in Alaska and Canada confirmed that atherosclerosis and myocardial infarction were present and not uncommon among the Inuit.[8] Furthermore, modern studies of Alaska Native populations using standardized adjudications show high rates of CHD and stroke, with risk clearly correlated with LDL-C and ApoB.[9] The apparent “low risk” in historical cohorts was also confounded by a shorter average lifespan and a high prevalence of infectious disease and accidental death, which prevented many individuals from surviving long enough to manifest clinical ASCVD.[8]
2. The Tsimane and Hadza: The Threshold Model in Action
The Tsimane forager-horticulturalists of the Bolivian Amazon have been documented as having the lowest prevalence of coronary artery disease of any population recorded to date.[11] This has been used by some to argue that systemic inflammation — which is high in the Tsimane due to chronic parasite burden — is not a primary driver of atherosclerosis.
A mechanistic review reconciles the Tsimane data perfectly with the lipid model. The Tsimane maintain a lifetime mean LDL-C of approximately 70 mg/dL and a mean blood pressure of 104/64 mmHg.[11] In this population, 85% of adults have a coronary artery calcium (CAC) score of zero, even into their 70s and 80s.[11] Rather than contradicting the lipid hypothesis, the Tsimane represent the “threshold model” of disease: atherosclerosis requires the presence of a sufficient concentration of atherogenic lipoproteins over time to initiate and drive plaque formation. Because the Tsimane maintain LDL-C levels well below the critical threshold throughout their entire lives, their arteries remain relatively free of plaque despite high levels of systemic inflammation (hs-CRP above 3.0 mg/L in 51% of the population).[11] This finding constitutes powerful evidence that ApoB-particle burden — not inflammation — is the primary initiator of the atherosclerotic process.
3. Familial Hypercholesterolemia and the “Healthy Survivor” Argument
Heterozygous familial hypercholesterolemia (HeFH) results in lifelong exposure to extreme LDL-C levels, typically above 190 mg/dL, and carries a 20-fold increased risk of premature CAD if left untreated.[15] Skeptics point to FH heterozygotes who survive into their 80s without medication as evidence that LDL-C is not causal.
The existence of “healthy survivors” in FH is explained by three primary mechanisms:
- Selection Bias: Early clinical cohorts were often identified through their family history of premature events, while those without events remained undiagnosed. Systematic screening now shows that the vast majority of untreated FH patients suffer early cardiovascular events.
- Protective Genetic Modifiers: Some FH individuals carry rare loss-of-function variants in the PCSK9 gene or other modifiers that significantly lower their ApoB count or improve LDL receptor recycling, effectively normalizing their risk to that of a non-FH individual.[19]
- Risk Factor Multipliers: Survivors often have an absence of other risk factors, such as smoking or hypertension, which would otherwise accelerate the entrapment of ApoB particles in the arterial wall.
Mendelian randomization studies and the dramatic success of early statin therapy in FH children further confirm the causality of lipids in this population.[15,17]
4. The French Paradox: A Temporal Illusion
The “French Paradox” — low CHD rates despite high saturated fat intake — was largely popularized in the 1990s as a rebuttal to the lipid-heart hypothesis.[23] However, detailed analyses by Law and Wald (1999) and others have effectively dismantled the paradox as a combination of under-reporting and a “time-lag” in dietary transition.
Consumption of animal fat and the resulting serum cholesterol elevations in France only reached Western norms in the late 1960s and 1970s, decades later than in Britain or the United States.[25] Because atherosclerosis is a cumulative disease resulting from decades of exposure, French mortality in the 1980s and 1990s reflected the lower lipid levels of the mid-20th century.[25] Additionally, standardized data from the MONICA project revealed that French physicians were historically less likely to certify deaths as CHD, often using codes for other cardiac pathologies. As the French population’s exposure time to high lipids has increased, their CHD mortality has predictably aligned with the lipid model.
5. Lean Mass Hyper-Responders (LMHR): A Developing Challenge
The “Lean Mass Hyper-Responder” (LMHR) phenotype describes lean individuals on ketogenic diets who experience extreme LDL-C spikes — often exceeding 200 mg/dL — while maintaining high HDL-C and low triglycerides.[27] Skeptics argue that these individuals are at low risk because they are metabolically healthy.
The KETO-CTA pilot study followed 100 LMHR individuals for 12 months using high-resolution coronary CT angiography.[27] While the study found that plaque progression was modest over one year and was not predicted by ApoB within that short window, this is consistent with the biology of atherosclerosis, which typically takes decades to manifest as measurable plaque in healthy individuals. Critically, baseline plaque burden did predict progression, suggesting that those with existing atherosclerosis remain vulnerable to high lipids regardless of their ketogenic state.[27] Falsification of the lipid hypothesis in this cohort would require a lifetime of exposure to such LDL levels without plaque formation — a condition not yet satisfied by current data.
6. The HDL Paradox and CETP Inhibitor Failures
The failure of multiple trials to reduce cardiovascular risk by raising HDL-C — specifically the CETP inhibitor trials (torcetrapib, dalcetrapib) and niacin trials (HPS2-THRIVE, AIM-HIGH) — has been used to argue that the lipid hypothesis is fundamentally flawed.[31,32,33]
In reality, these failures strengthen the ApoB-centered lipid hypothesis. They demonstrate that HDL-C is a “marker” rather than a “maker” of risk — a proxy for low triglyceride levels and favorable metabolic health rather than a causal therapeutic target. Conversely, every therapy that successfully lowers ApoB-containing particles — statins, ezetimibe, PCSK9 inhibitors, inclisiran, bempedoic acid — results in a proportional reduction in cardiovascular risk, regardless of mechanism of action.[35]
Section 3 — Mendelian Randomization and Causal Inference: The Genetic Proof
Mendelian randomization (MR) serves as a naturalized randomized controlled trial, using the random allocation of genetic alleles at conception to study the lifetime effects of an exposure. Because genetic variants are not affected by lifestyle or the presence of disease, MR circumvents the confounding and reverse causation that limit observational studies.
Establishing Causality Through Genetic Pathways
MR studies have unequivocally established that LDL-C and ApoB are causally related to ASCVD. The most powerful evidence comes from three genetic loci:
- PCSK9 Variants: Individuals with lifelong lower LDL-C due to loss-of-function variants in the PCSK9 gene exhibit an approximately 54% reduction in CHD risk per 38.7 mg/dL of LDL-C reduction — far exceeding the benefit seen in short-term statin trials.[1,2]
- NPC1L1 Variants: Genetic variants that mimic the action of ezetimibe show a consistent reduction in CHD risk proportional to the degree of LDL-C lowering, providing genetic validation for non-statin lipid-lowering therapies.[41]
- LPA Locus: MR data confirm that genetically elevated Lp(a) is an independent causal risk factor for ASCVD and calcific aortic valve stenosis, establishing a second ApoB-mediated causal pathway distinct from LDL.[3]
Table 2: Mendelian Randomization vs. Randomized Clinical Trials — CHD Risk Reduction per 38.7 mg/dL LDL-C Reduction
| LDL-Lowering Mechanism | MR Estimate (Lifetime) | RCT Estimate (5-Year) | Duration Difference |
| HMGCR (Statin target) | ~54.5% | ~22% | Lifetime vs. 5 years |
| PCSK9 (Evolocumab target) | ~54.5% | ~15% (2.2-yr follow-up) | Lifetime vs. 2.2 years |
| NPC1L1 (Ezetimibe target) | ~54.5% | ~15% (as add-on) | Lifetime vs. 7 years |
Data synthesized from Ference et al. (2012, 2017) and CTT meta-analyses.[1,2]
The three-fold greater benefit observed in MR studies compared to RCTs underscores the importance of cumulative exposure. Atherosclerosis is a result of “lipoprotein-years” (exposure × time). Lowering lipids early in life prevents the very initiation of the atherosclerotic process, whereas starting therapy in the sixth decade is an exercise in damage control on pre-existing plaque. These MR data represent the strongest possible causal evidence short of a decades-long randomized trial — which is ethically impossible to conduct.
Section 4 — Statin and PCSK9 Inhibitor Trial Evidence: A Critical Synthesis
The randomized controlled trial (RCT) evidence base for lipid-lowering therapy is among the most extensive in medical science. The Cholesterol Treatment Trialists’ (CTT) Collaboration meta-analysis of more than 26 trials involving over 170,000 participants demonstrated a 22% relative risk reduction in MACE for every 38.7 mg/dL (1 mmol/L) reduction in LDL-C, a result that held across all subgroups including sex, age, baseline LDL-C, and presence or absence of pre-existing cardiovascular disease.[35]
Countering the Pleiotropy Argument
A common heterodox argument suggests that statins reduce cardiovascular events via anti-inflammatory or other “pleiotropic” mechanisms independent of LDL-C lowering — implying that the benefits cannot be attributed to the lipid-lowering itself. This argument is falsified by the concordance of non-statin trials with the CTT regression line:
- IMPROVE-IT: Adding ezetimibe to simvastatin in 18,144 patients after acute coronary syndrome produced a further reduction in cardiovascular events that fell exactly on the CTT regression line, despite ezetimibe’s entirely different mechanism of action (intestinal cholesterol absorption inhibition via NPC1L1). Ezetimibe has no meaningful anti-inflammatory activity. Its benefit was purely proportional to the LDL-C reduction achieved.[38]
- FOURIER and ODYSSEY OUTCOMES: The PCSK9 inhibitors evolocumab and alirocumab reduced MACE proportionally to the absolute LDL-C reduction achieved, even in patients already on maximally-dosed high-intensity statin therapy — patients whose inflammatory markers were already suppressed.[36,37]
- FOURIER-OLE: Long-term open-label extension data demonstrated continued incremental risk reduction at ultra-low LDL-C levels (below 20 mg/dL), without any increase in adverse safety events, confirming the “lower is better” paradigm and ruling out a therapeutic floor effect or J-curve.[47]
Critics argue the benefit is “modest in absolute terms,” typically citing the 2–3 percentage point absolute risk reductions observed in trials of median 2.2 to 5 years. This critique misunderstands the statistical nature of atherosclerotic disease. When the same per-unit LDL-C reduction is modeled over a lifetime starting at age 40, the absolute benefit is an order of magnitude larger — precisely what the Mendelian randomization data confirm. The modest trial-level absolute risk reductions are a predictable artifact of short follow-up in middle-aged populations that are already decades into the atherosclerotic process.
Section 5 — Industry Interference and Motivated Skepticism: The Historical Record
5.1 The Sugar Research Foundation and Project 226
Historical analysis of internal industry documents has revealed that the Sugar Research Foundation (SRF) recognized as early as the 1950s that if Americans adopted low-fat diets, sucrose consumption would increase substantially. In 1965, the SRF funded “Project 226” — a literature review written by Harvard nutritionists D. Mark Hegsted and Fredrick Stare — that was intentionally designed to exonerate sucrose as a driver of CHD and focus the blame entirely on dietary fat and cholesterol.[48] The scientists involved received payments equivalent to approximately $50,000 in 2016 dollars and allowed the SRF to review drafts and select the literature to be included.[48] This successfully delayed the scientific consensus on sugar by decades and created a “fat vs. sugar” false dichotomy that heterodox figures continue to exploit today, claiming the lipid hypothesis was “founded on a lie” — a rhetorical misdirection that conflates the dietary fat debate with the entirely separate question of lipoprotein causality in ASCVD.
5.2 Egg, Dairy, and Meat Industry Funding Patterns
The pattern of industry funding influencing nutrition science continues in the present day. Between 2010 and 2019, 60% of research on eggs and blood cholesterol was sponsored by the egg industry.[54] While 86% of these studies actually found that eggs do raise LDL-C, half of them reported conclusions that were discordant with their own results — for example, claiming that the LDL-C increases were “not clinically significant” or were offset by changes in HDL-C.[54]
The dairy and meat industries have funded systematic reviews — notably the NutriRECS series published in Annals of Internal Medicine — that utilize GRADE methodology to classify all cohort-based nutritional evidence as “low certainty.”[57] While technically defensible on methodological grounds (long-term dietary RCTs are rarely feasible), this approach is operationally deployed to dismiss consistent, replicated risk signals and manufacture sufficient uncertainty to justify maintaining current high-meat dietary patterns. A systematic review examining the association of dairy industry funding with cardiovascular research findings found that industry-funded studies were significantly more likely to report neutral or favorable results for dairy products.[60]
Section 6 — The Commercial Ecosystem of Cholesterol Denial: High-Protein, Ketogenic, and Paleolithic Diet Industries
Beyond the food commodity industries described above, a distinct and highly organized commercial ecosystem has emerged that derives direct financial benefit from denying the atherogenic significance of LDL-C and ApoB. This ecosystem spans bestselling popular books, direct-to-consumer meat and supplement companies, paid influencer programs, podcast empires, and a small number of clinicians-turned-entrepreneurs who use the architecture of scientific discourse while systematically violating its methodological requirements. Understanding the financial structure of this ecosystem is essential to evaluating the credibility of its scientific claims.
6.1 The Core Commercial Logic
The central economic reality is straightforward: dietary guidelines limiting saturated fat and cholesterol directly restrict the market for red meat, processed meat, full-fat dairy products, and the high-protein supplement industry. By promoting a narrative that (a) saturated fat and dietary cholesterol are harmless or beneficial, (b) LDL-C is not a meaningful causal risk factor for ASCVD, and (c) high-protein, animal-fat-based diets are evolutionarily optimal, these commercial interests simultaneously expand sales and cultivate an ideologically committed consumer community that interprets scientific opposition as institutional corruption. The result is a self-reinforcing commercial-ideological machine that is extraordinarily resistant to empirical refutation.
6.2 The Ketogenic and Carnivore Diet Industry
The ketogenic and carnivore diet movement has produced a multi-hundred-million-dollar commercial ecosystem. Leading figures in this space — including Dave Feldman (creator of the LMHR concept), Paul Saladino (“CarnivoreMD”), Shawn Baker, Georgia Ede, and Ken Berry — simultaneously conduct or cite clinical research questioning LDL-C’s importance and operate businesses selling direct-to-consumer beef products, animal organ supplements, electrolyte formulas, online coaching subscriptions, and book publishing rights. Feldman’s “Citizen Science Foundation” and associated KETO-CTA study[27] have received significant attention for their one-year coronary CT data on lean mass hyper-responders. As discussed in Section 2.5, these data are preliminary, and their interpretation as exculpatory for high LDL-C is scientifically unjustified at this stage. The undisclosed financial relationships between the researchers and the ketogenic commercial ecosystem represent conflicts of interest that have not received adequate scrutiny in popular discourse.
The ketogenic diet industry’s anti-LDL argument rests on three claims, each of which fails under scrutiny:
- “Metabolic health negates LDL risk”: There is no randomized trial or Mendelian randomization study demonstrating that the presence of low triglycerides or high HDL neutralizes the atherogenic effect of elevated ApoB-particle number. The biological mechanism of subendothelial particle retention does not require metabolic dysfunction — it requires only a sufficient ApoB concentration gradient across the endothelial barrier.
- “The KETO-CTA showed LDL doesn’t predict plaque”: The study found that within-group variation in ApoB did not predict one-year plaque change in a 100-person cohort. This is far weaker than the claim made in popular reporting. The study’s own data showed that baseline plaque burden predicted progression — precisely consistent with the lipid model.
- “Ancestral humans ate high-fat diets without heart disease”: Autopsy data from mummified remains of ancient Egyptians, Peruvians, Puebloans, and Unangan individuals have documented atherosclerosis across multiple hunter-gatherer and agricultural populations — directly contradicting the “ancestral immunity” narrative promoted by Paleo and carnivore advocates.
6.3 The Paleolithic Diet Industry
The Paleo movement — commercialized through the work of Loren Cordain (whose books and related brand generate millions annually), Robb Wolf (Paleo Solution, NovOS supplement company), Mark Sisson (Primal Blueprint, Primal Kitchen brand acquired by Kraft Heinz), and Chris Kresser (Kresser Institute functional medicine training program) — has built substantial revenue streams through cookbooks, certification programs, supplement sales, and online subscription services. While the Paleo framework has contributed useful public health messaging around ultra-processed food avoidance, the industry has concurrently promoted a secondary narrative that is scientifically unfounded: that ancestral humans had uniformly low ASCVD burden and that high red meat and saturated fat consumption is therefore cardiovascular-neutral.
This narrative is contradicted by several lines of evidence. Hladik et al. and Thompson et al., examining CT imaging of mummified remains from four geographically and dietarily diverse ancient populations, documented probable or definite atherosclerosis in 47 of 137 mummies (34%), including in hunter-gatherer populations with no access to modern processed foods. The Paleo industry’s selective presentation of ancestral cardiovascular data to support high red-meat consumption constitutes motivated reasoning directly analogous to the food industry funding practices documented in Section 5.
6.4 The High-Protein Supplement Industry
The animal-derived protein supplement industry — encompassing whey protein, casein, collagen, beef protein isolate, and related products — has financial incentives to promote protein-centric dietary frameworks that emphasize high animal protein consumption and minimize concern about accompanying saturated fat and dietary cholesterol intake. Industry-funded research in this area has been used to advocate protein intakes well above established guidelines (often 1.6–2.4 g/kg/day or higher) while arguing that the associated increases in saturated fat intake are either metabolically irrelevant or actively beneficial due to effects on LDL particle size.
The LDL particle size argument — that large, “fluffy” LDL particles are benign even when elevated — has been formally examined and rejected in the scientific literature. Mendelian randomization studies using genetic instruments that specifically alter LDL particle size without changing LDL-C demonstrate that it is LDL particle number (i.e., ApoB) rather than size that mediates atherogenic risk. Large LDL particles are atherogenic at elevated concentrations; their larger size does not prevent subendothelial retention.
6.5 Gary Taubes and the Nutrition Science Initiative (NuSI)
Gary Taubes’ books Good Calories, Bad Calories (2007) and The Case Against Sugar (2016) have been enormously influential in establishing the popular narrative that the lipid hypothesis was fraudulently established by institutional suppression of the carbohydrate-insulin model of obesity. Taubes co-founded the Nutrition Science Initiative (NuSI) with Peter Attia in 2012, raising approximately $40 million from philanthropic donors to fund studies challenging the diet-heart hypothesis. The NuSI-funded studies generally failed to overturn the energy balance model of obesity, and the organization was effectively wound down by 2021. Nevertheless, Taubes’ argument — that Ancel Keys’ Seven Countries Study was cherry-picked and that the entire lipid hypothesis rests on a corrupt foundation — continues to circulate widely in the low-carbohydrate community.
The scientific problem with this framing is that it is entirely backward-looking. Even if one accepts every criticism of Keys’ original epidemiological work, the subsequent 50 years of Mendelian randomization, statin trials, PCSK9 inhibitor trials, and ezetimibe trials represent an entirely independent evidentiary base that did not exist when Keys was working and that conclusively establishes ApoB-particle causality through mechanisms that are immune to the confounding Taubes invokes against observational epidemiology.
6.6 Nina Teicholz and “The Big Fat Surprise”
Nina Teicholz’s 2014 book The Big Fat Surprise and her ongoing advocacy through The Nutrition Coalition constitute the most organized popular challenge to dietary fat guidance in the United States. A detailed fact-checking analysis by the BMJ found that Teicholz’s characterizations of the scientific literature contained multiple inaccuracies in the treatment of the evidence. Her work has been cited extensively by the meat and dairy industries and has been used to lobby for modifications to U.S. Dietary Guidelines. Teicholz’s central methodological error is identical to that of Taubes: she treats the historical epidemiological debate around dietary fat as dispositive for the question of lipoprotein causality in ASCVD, while refusing to engage with Mendelian randomization evidence — which is the most causally rigorous evidence type available — on the grounds that genetic studies are “not the same” as dietary intervention studies. This argument, while technically containing a kernel of truth (genetic LDL lowering and dietary LDL lowering may differ in magnitude), does not address the core point: both operate through the identical biological mechanism of reducing ApoB-particle infiltration of the arterial wall.
Section 7 — Organized Cholesterol Denialism: THINCS, Academic Contrarians, and Supplement Markets
Beyond commercially motivated skepticism, a distinct intellectual tradition of organized cholesterol denialism has emerged, characterized by coordinated advocacy for the position that LDL-C and ApoB have no causal role in ASCVD and, in extreme formulations, that elevated cholesterol may be protective. This movement is frequently cited in popular discourse as representing a legitimate scientific minority view, and it warrants systematic rebuttal.
7.1 The International Network of Cholesterol Skeptics (THINCS)
The International Network of Cholesterol Skeptics (THINCS) was founded by Danish physician Uffe Ravnskov, author of The Cholesterol Myths (2000) and Fat and Cholesterol Are Good for You (2009). THINCS maintains a manifesto — signed by approximately 100 physicians and researchers — asserting that the cholesterol hypothesis is unsupported by evidence, that cholesterol-lowering drugs provide no net benefit, and that high serum cholesterol is neutral or protective, particularly in elderly populations. The THINCS network publishes selectively in lower-tier journals, issues press releases to popular media, and maintains an active web presence that generates significant lay readership.
The principal scientific claims of THINCS fail under rigorous analysis on multiple grounds:
The “J-curve” in elderly populations: Ravnskov, David Diamond, and Malcolm Kendrick cite observational studies showing inverse or U-shaped associations between LDL-C and all-cause mortality in elderly populations as evidence that lower cholesterol is harmful. This argument commits two fundamental errors. First, it relies on reverse causation: in elderly or chronically ill individuals, low LDL-C is frequently a consequence of liver dysfunction, malignancy, cardiac cachexia, or chronic inflammation — not a cause of mortality. When this confounding is controlled by excluding early deaths (“leave-one-out” analyses) or by using Mendelian randomization, the apparently protective association disappears entirely. Second, all-cause mortality and ASCVD-specific mortality are different endpoints; even if LDL-C were inversely associated with all-cause mortality in frail elderly individuals (which is entirely explicable by reverse causation), this would not bear on the causal relationship between ApoB and ASCVD that operates across a decades-long biological timescale.
“Statins are harmful and their benefits are exaggerated”: Ravnskov, John Abramson, and Kendrick have argued in peer-reviewed publications and popular books that statin benefits are overstated through relative rather than absolute risk framing, and that statins cause widespread muscle damage, cognitive impairment, and metabolic harm. The absolute-versus-relative risk framing argument is a general concern in medical communication and does not address the underlying biology. The cognitive impairment claim was amplified following an FDA label update in 2012 but has not been confirmed in any prospective, blinded, randomized trial. The largest meta-analyses of statin safety data — encompassing hundreds of thousands of patient-years of follow-up — confirm that serious adverse events are uncommon and that statin therapy is well-tolerated in the vast majority of patients.
“The lipid hypothesis was not proven in women”: Some THINCS-affiliated arguments claim that lipid lowering has not been shown to reduce cardiovascular events in women. This is directly contradicted by CTT collaboration subgroup analyses, the JUPITER trial (rosuvastatin in primary prevention, demonstrating consistent benefit in women), and Mendelian randomization data that show proportional ASCVD risk reduction in females across all genetic LDL-lowering instruments.
7.2 The “Cholesterol Is Vital” Non-Sequitur
A rhetorical device common to both THINCS and the high-protein diet community is the invocation of cholesterol’s essential biological functions — steroid hormone synthesis, cell membrane integrity, bile acid production, neuronal function, vitamin D synthesis — to imply that elevated circulating levels of cholesterol-carrying lipoproteins are therefore beneficial or benign. This represents a categorical logical error that Zoë Harcombe deploys with particular frequency on her website and in peer-reviewed correspondence.[67]
Cholesterol is essential at the cellular level, and intracellular cholesterol biosynthesis is exquisitely regulated by the mevalonate pathway. The pathological entity in ASCVD is not cholesterol per se but ApoB-containing lipoproteins — macromolecular particles capable of crossing the endothelial barrier and being retained within the subendothelial space through interaction with proteoglycans. Neonates and other primates maintain plasma LDL-C in the range of 40–70 mg/dL with entirely normal steroid hormone levels, cell membrane composition, and neurological function, demonstrating that cholesterol’s essential biological functions are fully served at concentrations far below those associated with ASCVD risk in Western populations. The argument that “cholesterol is vital” is thus a biological truism that has no bearing on the pathological effects of elevated plasma ApoB-particle burden.
7.3 The Alternative Medicine and Supplement Industry Interface
The cholesterol denialism ecosystem intersects substantially with the functional medicine and alternative health supplement industries. By characterizing statins as toxic agents whose benefits are systematically overstated by pharmaceutical company-funded research, this ecosystem creates demand for proprietary “natural” alternatives: red yeast rice, berberine, omega-3 concentrates, CoQ10 products (marketed to address the widely promoted but poorly substantiated claim that statins deplete CoQ10 and cause fatigue), polyphenol extracts, plant sterol supplements, and various combination products sold by functional medicine practitioners and direct-to-consumer online pharmacies.
The commercial irony of this position is pointed: red yeast rice contains monacolin K — the chemically identical molecule to lovastatin — at pharmacologically active concentrations. The anti-statin argument is therefore not fundamentally about mechanism but about directing revenue from generic pharmaceutical products toward higher-margin proprietary supplements sold outside the regulatory framework that governs prescription drugs. Practitioners promoting this substitution do their patients a disservice on two levels: they deny them a therapy with an extensive, validated safety and efficacy record, and they replace it with a preparation whose monacolin K content varies up to 100-fold between brands and is not subject to pharmacovigilance.
7.4 The “Research Corruption” Meta-Argument and Its Limits
Perhaps the most rhetorically powerful tool in the cholesterol denial ecosystem is the “research corruption” meta-argument: the claim that the entire evidence base for lipid-lowering is compromised by pharmaceutical industry funding of statin trials, regulatory capture of guideline-writing bodies, and financial relationships between academic lipidologists and PCSK9 inhibitor manufacturers. This argument is not entirely without foundation — conflicts of interest in cardiovascular medicine are real and warrant ongoing scrutiny. However, as deployed in the denialist literature, it is used to dismiss entire categories of evidence rather than to improve its quality.
The fundamental problem with the research corruption argument is that it cannot account for the Mendelian randomization evidence, which is entirely independent of pharmaceutical funding, is generated from national biobanks and population registries using publicly available genetic data, and produces results in complete concordance with the trial evidence. The individuals with loss-of-function PCSK9 variants who have 54% lower lifetime CHD risk were not enrolled in a pharmaceutical trial and received no intervention — their protection derives from a genetic variant that was present at conception, before any pharmaceutical company existed to fund research about it. Dismissing this evidence requires a conspiratorial framework that has no foundation in the scientific record.
Section 8 — The Threshold Model and Evolutionary Context
A synthesis of the Tsimane, Hadza, and pre-industrial data into a coherent model suggests that atherosclerosis is not an inevitable consequence of aging but a condition of high-lipoprotein exposure sustained over time.
The Genetic Normative Environment
For most of human evolutionary history, LDL-C levels were likely in the range of 40 to 70 mg/dL — similar to those seen in modern non-human primates and human neonates.[11] In this range, ApoB particles are present at such a low concentration that their rate of infiltration and subendothelial entrapment is negligible. Subclinical atherosclerosis, however, begins early in high-lipoprotein environments. Autopsy data from the Bogalusa Heart Study and CARDIA show that aortic fatty streaks and fibrous plaques begin forming in the second decade of life and are strongly correlated with LDL-C and VLDL levels measured in childhood.[69,70] This finding has profound implications: the atherosclerotic clock begins before adulthood, and the cumulative exposure integral — ApoB concentration × time, or “ApoB-years” — is the primary determinant of eventual clinical risk.
Table 3: Cumulative ApoB Exposure and Plaque Development — The ApoB-Year Model
| Population / Context | Lifetime ApoB Target (approx.) | Onset of Raised Fibrous Plaque | Clinical Event Risk |
| Pre-Industrial (Tsimane) | ~50 mg/dL | Rare/absent | Near-zero |
| Modern Western Average | ~90–110 mg/dL | 2nd–3rd Decade | High (Age 50+) |
| Heterozygous FH (untreated) | >150 mg/dL | Childhood | Extreme (Age 30+) |
Synthesized from Bogalusa Heart Study, Tsimane cohort data, and FH registry data.[11,15,69,70]
This threshold model predicts that to avoid meaningful atherosclerosis, the lifetime average ApoB should be maintained below approximately 50–60 mg/dL. Once the cumulative ApoB-year exposure exceeds a critical threshold, the burden of subendothelial plaque reaches the point at which vulnerable plaque rupture, thrombosis, and acute coronary events become increasingly probable. The therapeutic implication is that earlier treatment initiation produces disproportionately greater lifetime benefit — consistent with every line of evidence from MR, RCTs, and population data.
Conclusion: The Preponderance of Evidence and Falsification Criteria
The totality of evidence from biochemistry, large prospective cohorts, Mendelian randomization, and randomized controlled trials unequivocally establishes that ApoB-containing lipoproteins are the causal agents of ASCVD.[1,2] The scientific consensus is not genuinely “uncertain”; rather, uncertainty has been systematically manufactured by a coalition of interests: (1) the sugar and refined carbohydrate industries, which redirected scientific attention away from sucrose in the 1960s[48]; (2) the egg, dairy, and meat commodity industries, which continue to fund research designed to minimize the perceived significance of dietary lipid-raising[54,57,60]; (3) the high-protein, ketogenic, carnivore, and Paleolithic diet commercial ecosystem, which has direct revenue interest in denying the atherogenic role of saturated fat and red meat consumption[27]; (4) the organized THINCS cholesterol denial network and its associated alternative medicine markets; and (5) a small group of academic contrarians whose arguments consistently avoid engagement with Mendelian randomization evidence — the most causally rigorous data type available.
Falsification Criteria
To scientifically falsify the atherogenic lipoprotein hypothesis, a researcher would need to demonstrate at least one of the following:
- Identify a population with high lifetime ApoB levels (above 100 mg/dL sustained) that does not develop coronary artery calcium or atherosclerosis even in the eighth or ninth decade of life. The LMHR cohort is the only current candidate, but current data span only 12 months in 100 individuals — wholly insufficient to falsify a hypothesis about decades-long biological accumulation.
- Conduct a randomized trial in which a substantial, isolated reduction in ApoB-containing particles fails to produce a proportional reduction in cardiovascular events over a minimum of five years. No such trial has been conducted, and such a trial would face serious ethical barriers given the existing evidence.
To date, every study conducted has further confirmed the model. The preponderance of evidence supports a “lower is better, and lower for longer is best” paradigm, with ApoB serving as the most accurate quantitative metric for the atherogenic particle burden driving disease. The argument that high LDL-C or ApoB is irrelevant to ASCVD is not a scientific minority view competing on equal epistemic footing with the consensus — it is a commercially motivated claim that has failed every rigorous empirical test to which it has been subjected.
References
All references are peer-reviewed journal publications.
- Ference BA, Ginsberg HN, Graham I, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2017;38(32):2459-2472. doi:10.1093/eurheartj/ehx144
- Ference BA, Yoo W, Alesh I, et al. Effect of long-term exposure to lower low-density lipoprotein cholesterol beginning early in life on the risk of coronary heart disease: a Mendelian randomization analysis. J Am Coll Cardiol. 2012;60(25):2631-2639. doi:10.1016/j.jacc.2012.09.017
- Doherty S, Hernandez S, Rikhi R, et al. Lipoprotein(a) as a Causal Risk Factor for Cardiovascular Disease. Curr Cardiovasc Risk Rep. 2025;19(1):8. doi:10.1007/s12170-025-00760-1
- Du Y, Ding W, Ye Z, et al. Impact of LDL-C and Apolipoprotein B Level Discordance and associated Lipoprotein Particle Alterations on Cardiovascular Outcomes in a large primary prevention population. Eur J Prev Cardiol. Published online November 24, 2025. doi:10.1093/eurjpc/zwaf750
- Borén J, Williams KJ. The central role of arterial retention of cholesterol-rich apolipoprotein-B-containing lipoproteins in the pathogenesis of atherosclerosis: a triumph of simplicity. Curr Opin Lipidol. 2016;27(5):473-483. doi:10.1097/MOL.0000000000000330
- Sniderman AD, Thanassoulis G, Glavinovic T, et al. Apolipoprotein B Particles and Cardiovascular Disease: A Narrative Review. JAMA Cardiol. 2019;4(12):1287-1295. doi:10.1001/jamacardio.2019.3780
- Bang HO, Dyerberg J, Hjøorne N. The composition of food consumed by Greenland Eskimos. Acta Med Scand. 1976;200(1-2):69-73. doi:10.1111/j.0954-6820.1976.tb08198.x
- Fodor JG, Helis E, Yazdekhasti N, Vohnout B. “Fishing” for the origins of the “Eskimos and heart disease” story: facts or wishful thinking?. Can J Cardiol. 2014;30(8):864-868. doi:10.1016/j.cjca.2014.04.007
- Howard BV, Devereux RB, Cole SA, et al. A genetic and epidemiologic study of cardiovascular disease in Alaska natives (GOCADAN): design and methods. Int J Circumpolar Health. 2005;64(3):206-221. doi:10.3402/ijch.v64i3.17985
- Howard BV, Robbins DC, Sievers ML, et al. LDL cholesterol as a strong predictor of coronary heart disease in diabetic individuals with insulin resistance and low LDL: The Strong Heart Study. Arterioscler Thromb Vasc Biol. 2000;20(3):830-835. doi:10.1161/01.atv.20.3.830
- Kaplan H, Thompson RC, Trumble BC, et al. Coronary atherosclerosis in indigenous South American Tsimane: a cross-sectional cohort study. Lancet. 2017;389(10080):1730-1739. doi:10.1016/S0140-6736(17)30752-3
- Gurven M, Kaplan H, Winking J, Finch C, Crimmins EM. Aging and inflammation in two epidemiological worlds. J Gerontol A Biol Sci Med Sci. 2008;63(2):196-199. doi:10.1093/gerona/63.2.196
- Thompson RC, Allam AH, Lombardi GP, et al. Atherosclerosis across 4000 years of human history: the Horus study of four ancient populations.
- Trumble BC, Stieglitz J, Blackwell AD, et al. Apolipoprotein E4 is associated with improved cognitive function in Amazonian forager-horticulturalists with a high parasite burden. FASEB J. 2017;31(4):1508-1515. doi:10.1096/fj.201601084R
- Nordestgaard BG, Chapman MJ, Humphries SE, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013;34(45):3478-90a. doi:10.1093/eurheartj/eht273
- Defesche JC, Gidding SS, Harada-Shiba M, Hegele RA, Santos RD, Wierzbicki AS. Familial hypercholesterolaemia. Nat Rev Dis Primers. 2017;3:17093. Published 2017 Dec 7. doi:10.1038/nrdp.2017.93
- Wiegman A, Gidding SS, Watts GF, et al. Familial hypercholesterolaemia in children and adolescents: gaining decades of life by optimizing detection and treatment. Eur Heart J. 2015;36(36):2425-2437. doi:10.1093/eurheartj/ehv157
- Besseling J, Hovingh GK, Huijgen R, Kastelein JJP, Hutten BA. Statins in Familial Hypercholesterolemia: Consequences for Coronary Artery Disease and All-Cause Mortality. J Am Coll Cardiol. 2016;68(3):252-260. doi:10.1016/j.jacc.2016.04.054
- Abifadel M, Varret M, Rabès JP, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia.
- Hovingh GK, Davidson MH, Kastelein JJ, O’Connor AM. Diagnosis and treatment of familial hypercholesterolaemia. Eur Heart J. 2013;34(13):962-971. doi:10.1093/eurheartj/eht015
- Raal FJ, Honarpour N, Blom DJ, et al. Inhibition of PCSK9 with evolocumab in homozygous familial hypercholesterolaemia (TESLA Part B): a randomised, double-blind, placebo-controlled trial. Lancet. 2015;385(9965):341-350. doi:10.1016/S0140-6736(14)61374-X
- Ibrahim S, Reeskamp LF, de Goeij JN, et al. Beyond early LDL cholesterol lowering to prevent coronary atherosclerosis in familial hypercholesterolaemia. Eur J Prev Cardiol. 2024;31(7):892-900. doi:10.1093/eurjpc/zwae028
- Law M, Wald N. Why heart disease mortality is low in France: the time lag explanation. BMJ. 1999;318(7196):1471-1476. doi:10.1136/bmj.318.7196.1471
- Simini B. Serge Renaud: from French paradox to Cretan miracle. Lancet. 2000;355(9197):48. doi:10.1016/S0140-6736(05)71990-5
- Ferrières J. The French paradox: lessons for other countries. Heart. 2004;90(1):107-111. doi:10.1136/heart.90.1.107
- GBD 2023 Disease and Injury and Risk Factor Collaborators. Burden of 375 diseases and injuries, risk-attributable burden of 88 risk factors, and healthy life expectancy in 204 countries and territories, including 660 subnational locations, 1990-2023: a systematic analysis for the Global Burden of Disease Study 2023. Lancet. 2025;406(10513):1873-1922. doi:10.1016/S0140-6736(25)01637-X
- Norwitz NG, Feldman D, Soto-Mota A, et al. The impact of sustained LDL-C elevation on coronary plaque progression: primary results from the KETO-CTA study. medRxiv. 2026. doi:10.64898/2026.01.15.26343955v1
- Norwitz NG, Soto-Mota A, Feldman D, Parpos S, Budoff M. Case Report: Hypercholesterolemia “Lean Mass Hyper-Responder” Phenotype Presents in the Context of a Low Saturated Fat Carbohydrate-Restricted Diet. Front Endocrinol (Lausanne). 2022;13:830325. Published 2022 Apr 14. doi:10.3389/fendo.2022.830325
- Borén J, Chapman MJ, Krauss RM, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2020;41(24):2313-2330. doi:10.1093/eurheartj/ehz962
- Packard C, Caslake M, Shepherd J. The role of small, dense low density lipoprotein (LDL): a new look. Int J Cardiol. 2000;74 Suppl 1:S17-S22. doi:10.1016/s0167-5273(99)00107-2
- Barter PJ, Caulfield M, Eriksson M, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357(21):2109-2122. doi:10.1056/NEJMoa0706628
- HPS2-THRIVE Collaborative Group, Landray MJ, Haynes R, et al. Effects of extended-release niacin with laropiprant in high-risk patients. N Engl J Med. 2014;371(3):203-212. doi:10.1056/NEJMoa1300955
- Schwartz GG, Olsson AG, Abt M, et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. 2012;367(22):2089-2099. doi:10.1056/NEJMoa1206797
- AIM-HIGH Investigators, Boden WE, Probstfield JL, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365(24):2255-2267. doi:10.1056/NEJMoa1107579
- Cholesterol Treatment Trialists’ (CTT) Collaboration, Baigent C, Blackwell L, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376(9753):1670-1681. doi:10.1016/S0140-6736(10)61350-5
- Sabatine MS, Giugliano RP, Keech AC, et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N Engl J Med. 2017;376(18):1713-1722. doi:10.1056/NEJMoa1615664
- Schwartz GG, Steg PG, Szarek M, et al. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N Engl J Med. 2018;379(22):2097-2107. doi:10.1056/NEJMoa1801174
- Cannon CP, Blazing MA, Giugliano RP, et al. Ezetimibe Added to Statin Therapy after Acute Coronary Syndromes. N Engl J Med. 2015;372(25):2387-2397. doi:10.1056/NEJMoa1410489
- Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359(21):2195-2207. doi:10.1056/NEJMoa0807646
- Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354(12):1264-1272. doi:10.1056/NEJMoa054013
- Ference BA, Majeed F, Penumetcha R, Flack JM, Brook RD. Effect of naturally random allocation to lower low-density lipoprotein cholesterol on the risk of coronary heart disease mediated by polymorphisms in NPC1L1, HMGCR, or both: a 2 × 2 factorial Mendelian randomization study. J Am Coll Cardiol. 2015;65(15):1552-1561. doi:10.1016/j.jacc.2015.02.020
- Burgess S, Ference BA, Staley JR, et al. Association of LPA Variants With Risk of Coronary Disease and the Implications for Lipoprotein(a)-Lowering Therapies: A Mendelian Randomization Analysis. JAMA Cardiol. 2018;3(7):619-627. doi:10.1001/jamacardio.2018.1470
- Patel AP, Wang (汪敏先) M, Pirruccello JP, et al. Lp(a) (Lipoprotein[a]) Concentrations and Incident Atherosclerotic Cardiovascular Disease: New Insights From a Large National Biobank. Arterioscler Thromb Vasc Biol. 2021;41(1):465-474. doi:10.1161/ATVBAHA.120.315291
- Clarke R, Peden JF, Hopewell JC, et al. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med. 2009;361(26):2518-2528. doi:10.1056/NEJMoa0902604
- Ference BA, Kastelein JJP, Ray KK, et al. Association of Triglyceride-Lowering LPL Variants and LDL-C-Lowering LDLR Variants With Risk of Coronary Heart Disease. JAMA. 2019;321(4):364-373. doi:10.1001/jama.2018.20045
- Sabatine MS, Giugliano RP, Wiviott SD, et al. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372(16):1500-1509. doi:10.1056/NEJMoa1500858
- O’Donoghue ML, Giugliano RP, Wiviott SD, et al. Long-Term Evolocumab in Patients With Established Atherosclerotic Cardiovascular Disease. Circulation. 2022;146(15):1109-1119. doi:10.1161/CIRCULATIONAHA.122.061620
- Kearns CE, Schmidt LA, Glantz SA. Sugar Industry and Coronary Heart Disease Research: A Historical Analysis of Internal Industry Documents.
- Kearns CE, Apollonio D, Glantz SA. Sugar industry sponsorship of germ-free rodent studies linking sucrose to hyperlipidemia and cancer: An historical analysis of internal documents. PLoS Biol. 2017;15(11):e2003460. Published 2017 Nov 21. doi:10.1371/journal.pbio.2003460
- Nestle M. Food company sponsorship of nutrition research and professional activities: a conflict of interest?. Public Health Nutr. 2001;4(5):1015-1022. doi:10.1079/phn2001253
- Stuckler D, Nestle M. Big food, food systems, and global health. PLoS Med. 2012;9(6):e1001242. doi:10.1371/journal.pmed.1001242
- Nestle M. Food Industry Funding of Nutrition Research: The Relevance of History for Current Debates. JAMA Intern Med. 2016;176(11):1685-1686. doi:10.1001/jamainternmed.2016.5400
- Moodie R, Stuckler D, Monteiro C, et al. Profits and pandemics: prevention of harmful effects of tobacco, alcohol, and ultra-processed food and drink industries. Lancet. 2013;381(9867):670-679. doi:10.1016/S0140-6736(12)62089-3
- Barnard ND, Long MB, Ferguson JM, Flores R, Kahleova H. Industry Funding and Cholesterol Research: A Systematic Review. Am J Lifestyle Med. 2019;15(2):165-172. Published 2019 Dec 11. doi:10.1177/1559827619892198
- Zhong VW, Van Horn L, Cornelis MC, et al. Associations of Dietary Cholesterol or Egg Consumption With Incident Cardiovascular Disease and Mortality. JAMA. 2019;321(11):1081-1095. doi:10.1001/jama.2019.1572
- Shin JY, Xun P, Nakamura Y, He K. Egg consumption in relation to risk of cardiovascular disease and diabetes: a systematic review and meta-analysis. Am J Clin Nutr. 2013;98(1):146-159. doi:10.3945/ajcn.112.051318
- Johnston BC, Zeraatkar D, Han MA, et al. Unprocessed Red Meat and Processed Meat Consumption: Dietary Guideline Recommendations From the Nutritional Recommendations (NutriRECS) Consortium. Ann Intern Med. 2019;171(10):756-764. doi:10.7326/M19-1621
- Zeraatkar D, Johnston BC, Bartoszko J, et al. Effect of Lower Versus Higher Red Meat Intake on Cardiometabolic and Cancer Outcomes: A Systematic Review of Randomized Trials. Ann Intern Med. 2019;171(10):721-731. doi:10.7326/M19-0622
- Micha R, Michas G, Mozaffarian D. Unprocessed red and processed meats and risk of coronary artery disease and type 2 diabetes–an updated review of the evidence. Curr Atheroscler Rep. 2012;14(6):515-524. doi:10.1007/s11883-012-0282-8
- Guo J, Astrup A, Lovegrove JA, Gijsbers L, Givens DI, Soedamah-Muthu SS. Milk and dairy consumption and risk of cardiovascular diseases and all-cause mortality: dose-response meta-analysis of prospective cohort studies. Eur J Epidemiol. 2017;32(4):269-287. doi:10.1007/s10654-017-0243-1
- Willett WC, Ludwig DS. Milk and Health. N Engl J Med. 2020;382(7):644-654. doi:10.1056/NEJMra1903547
- Satija A, Bhupathiraju SN, Spiegelman D, et al. Healthful and Unhealthful Plant-Based Diets and the Risk of Coronary Heart Disease in U.S. Adults. J Am Coll Cardiol. 2017;70(4):411-422. doi:10.1016/j.jacc.2017.05.047
- Krauss RM, Blanche PJ, Rawlings RS, Fernstrom HS, Williams PT. Separate effects of reduced carbohydrate intake and weight loss on atherogenic dyslipidemia. Am J Clin Nutr. 2006;83(5):1025-1205. doi:10.1093/ajcn/83.5.1025
- Siri-Tarino PW, Sun Q, Hu FB, Krauss RM. Meta-analysis of prospective cohort studies evaluating the association of saturated fat with cardiovascular disease. Am J Clin Nutr. 2010;91(3):535-546. doi:10.3945/ajcn.2009.27725
- Keys A, Menotti A, Karvonen MJ, et al. The diet and 15-year death rate in the seven countries study. Am J Epidemiol. 1986;124(6):903-915. doi:10.1093/oxfordjournals.aje.a114480
- Menotti A, Puddu PE. How the Seven Countries Study contributed to the definition and development of the Mediterranean diet concept: a 50-year journey. Nutr Metab Cardiovasc Dis. 2015;25(3):245-252. doi:10.1016/j.numecd.2014.12.001
- Ravnskov U, Diamond DM, Hama R, et al. Lack of an association or an inverse association between low-density-lipoprotein cholesterol and mortality in the elderly: a systematic review. BMJ Open. 2016;6(6):e010401. Published 2016 Jun 12. doi:10.1136/bmjopen-2015-010401
- Ference BA, Bhatt DL, Catapano AL, et al. Association of Genetic Variants Related to Combined Exposure to Lower Low-Density Lipoproteins and Lower Systolic Blood Pressure With Lifetime Risk of Cardiovascular Disease. JAMA. 2019;322(14):1381-1391. doi:10.1001/jama.2019.14120
- Berenson GS, Srinivasan SR, Bao W, Newman WP 3rd, Tracy RE, Wattigney WA. Association between multiple cardiovascular risk factors and atherosclerosis in children and young adults. The Bogalusa Heart Study. N Engl J Med. 1998;338(23):1650-1656. doi:10.1056/NEJM199806043382302
- Pletcher MJ, Bibbins-Domingo K, Liu K, et al. Nonoptimal lipids commonly present in young adults and coronary calcium later in life: the CARDIA (Coronary Artery Risk Development in Young Adults) study. Ann Intern Med. 2010;153(3):137-146. doi:10.7326/0003-4819-153-3-201008030-00004
- McGill HC Jr, McMahan CA, Herderick EE, Malcom GT, Tracy RE, Strong JP. Origin of atherosclerosis in childhood and adolescence. Am J Clin Nutr. 2000;72(5 Suppl):1307S-1315S. doi:10.1093/ajcn/72.5.1307s
- Stary HC, Chandler AB, Glagov S, et al. A definition of initial, fatty streak, and intermediate lesions of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Arterioscler Thromb. 1994;14(5):840-856. doi:10.1161/01.atv.14.5.840