A Comprehensive Evaluation of Biomarkers in Cardiovascular Risk Stratification
I. Introduction: The Paradigm Shift
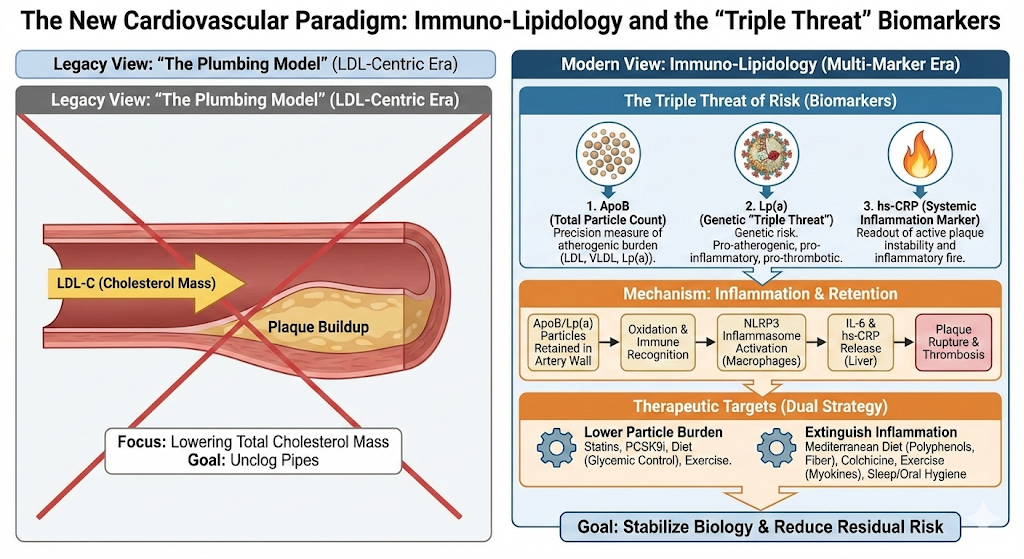
For decades, preventive cardiology has been anchored by a single, powerful concept: the “lipid hypothesis.” We operated under the assumption that cholesterol accumulation—specifically LDL-C—was the primary driver of atherosclerotic cardiovascular disease. The clinical directive was straightforward: push LDL-C down, and heart attack risk will follow. While this approach, largely driven by statin therapy, has undeniably saved millions of lives, we now know it is an incomplete strategy.
Even when patients achieve aggressive LDL targets, a significant “residual risk” remains. We see this clinically every day: patients with “perfect” cholesterol levels who still suffer heart attacks. This persistence of risk has forced a re-evaluation of the disease model. We are moving away from viewing atherosclerosis as a passive “plumbing” problem—pipes clogged by grease—toward understanding it as immuno-lipidology. In this view, plaque is not just debris; it is a chronic, maladaptive inflammatory response to lipoprotein retention.
Two major developments—the 30-year follow-up of the Women’s Health Study (published in the New England Journal of Medicine in late 2024) [1] and the 2025 Scientific Statement from the American College of Cardiology [2]—have fundamentally reshuffled our risk hierarchy. These reports suggest that High-Sensitivity C-Reactive Protein (hs-CRP), a marker of systemic inflammation, may actually outperform LDL-C in predicting long-term outcomes for many patients. At the same time, the reliance on LDL-C as a measure of particle burden is being rightly challenged by Apolipoprotein B (ApoB).
This report analyzes this shift, moving beyond the headlines to the pathophysiology, and provides an evidence-based roadmap for managing this “triple threat” of risk: particle count, genetic susceptibility, and inflammation.
II. The Mechanism: Why Inflammation Matters
To understand why a liver protein (CRP) might predict a heart attack better than cholesterol, we have to look at the vessel wall. The endothelium is not a passive lining; it is biologically active.
2.1 Retention and Oxidation
The process starts with retention. Apolipoprotein B-containing particles (LDL, VLDL, Lp(a)) migrate into the sub-endothelial space. Crucially, they get stuck. Once trapped, these particles oxidize. The immune system does not recognize Oxidized LDL (OxLDL) as “self”; it perceives it as a pathogen, similar to a bacterium.
2.2 The NLRP3 Inflammasome
This triggers the innate immune response. Macrophages rush in to engulf the oxidized lipids, becoming “foam cells.” This ingestion activates the NLRP3 inflammasome, a molecular complex that acts as a siren. It churns out Interleukin-1β (IL-1β), which in turn stimulates Interleukin-6 (IL-6). When IL-6 reaches the liver, it triggers the production of C-Reactive Protein (CRP).
Consequently, an elevated hs-CRP isn’t just a vague sign of “swelling.” It is a downstream readout of active plaque instability. Cholesterol builds the plaque, but inflammation is what weakens the cap, leading to rupture and thrombosis. This explains the data [3]: you can have a large, stable plaque (high cholesterol, low inflammation) that remains asymptomatic, or a smaller, inflamed plaque that ruptures and kills.
III. The Biomarkers: A Modern Hierarchy
We need to evaluate the three key players in this landscape: hs-CRP, ApoB, and Lipoprotein(a).
3.1 High-Sensitivity C-Reactive Protein (hs-CRP)
Standard CRP tests detect acute infections. For cardiovascular risk, we need high-sensitivity assays that detect low-grade inflammation (typically 0.5 to 10 mg/L).
- Low Risk: < 1.0 mg/L
- High Risk: > 3.0 mg/L
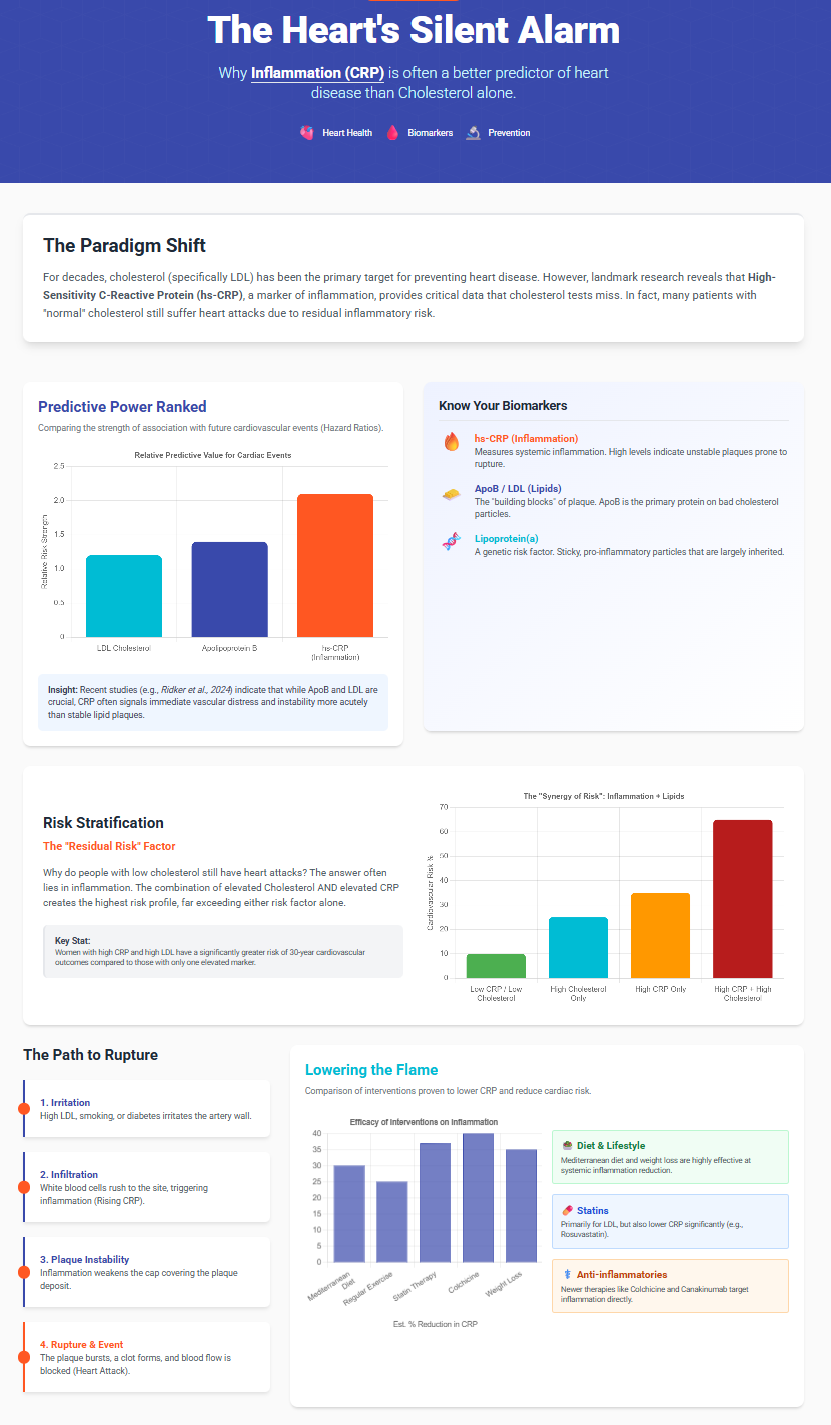
The 2024 Ridker study provided a sobering look at long-term risk. In nearly 28,000 women followed for 30 years, hs-CRP was a stronger predictor of future events than LDL-C [1]. The hazard ratio for the highest quintile of hs-CRP was 1.70, compared to 1.36 for LDL-C. This suggests that for long-term prognosis, knowing a patient’s inflammatory status is non-negotiable.
This aligns with previous data from the JUPITER trial [5], which showed that rosuvastatin reduced events significantly in people with normal LDL but high hs-CRP. Notably, the greatest benefit was seen in those who achieved lower hs-CRP levels, suggesting a strong association between reducing inflammation and survival.
3.2 Apolipoprotein B (ApoB)
LDL-C measures the mass of cholesterol, but the arterial wall cares about the number of particles. ApoB provides a 1:1 count of every atherogenic particle in circulation.
In patients with metabolic syndrome, obesity, or insulin resistance, we often see discordance: the LDL-C is normal, but the ApoB is sky-high because the cholesterol is carried in many small, dense particles. These small particles are more easily trapped in the arterial wall. This is why ApoB is a superior metric for estimating true lipoprotein burden [6, 7].
3.3 Lipoprotein(a) [Lp(a)]
Lp(a) is often called the “genetic triple threat.” It is an LDL particle attached to an apo(a) tail. It promotes atherosclerosis (via cholesterol), inflammation (it carries oxidized phospholipids) [8], and thrombosis (it mimics plasminogen, inhibiting clot breakdown).
Unlike the other markers, Lp(a) is 80-90% genetic and barely moves with diet or exercise. It represents a fixed baseline risk. The new guidelines recommend every adult test this once to identify “hidden” risk that standard panels miss [2].
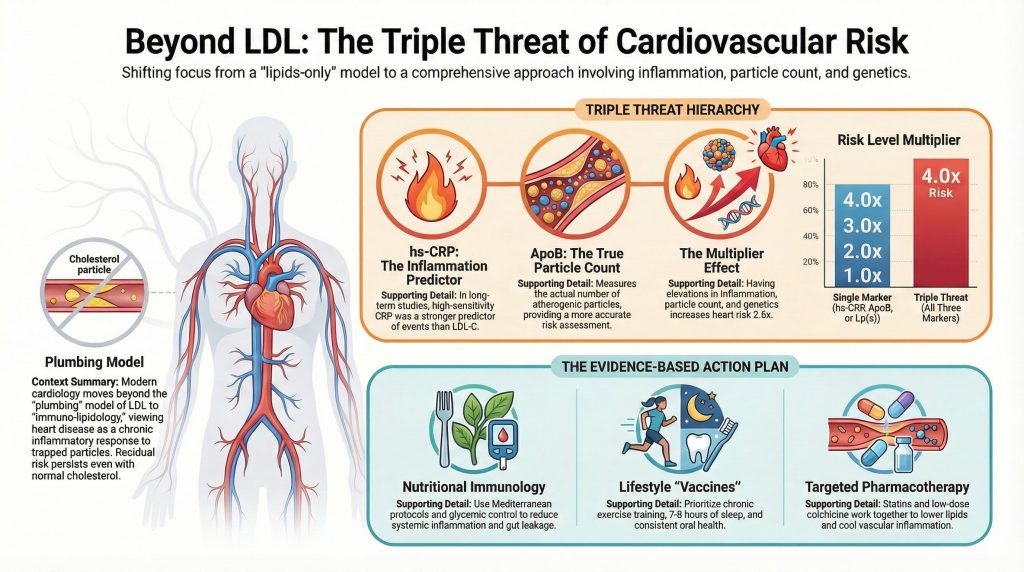
IV. Stratifying the “Triple Threat”
The most important takeaway from the 2024/2025 data is not that we should swap LDL for CRP, but that the risks are additive.
The Ridker data showed that while single elevations are bad, the combination is exponential [1].
- Single Marker Elevated: ~1.2x Risk
- All Three Elevated: ~2.6x Risk
The most dangerous patient is the one with high particle count (ApoB), high genetic susceptibility (Lp(a)), and active inflammation (hs-CRP).
V. From Theory to Therapy: Reducing Inflammatory Risk
If hs-CRP is high (>2.0 mg/L), “watch and wait” is no longer appropriate. We need a multimodal approach to cool the system.
5.1 Nutritional Immunology
Diet is our primary lever. The Mediterranean diet remains the gold standard, not just for lipids, but for inflammation [9].
- Mechanism: It’s not just about “healthy fats.” Extra virgin olive oil contains oleocanthal, a natural anti-inflammatory. High fiber intake feeds gut bacteria that produce butyrate, which strengthens the gut barrier and prevents bacterial toxins (LPS) from leaking into the bloodstream—a major cause of systemic inflammation.
- Glycemic Control: Insulin resistance is a pro-inflammatory state. Minimizing glucose spikes and eliminating ultra-processed foods (which often contain gut-disrupting emulsifiers) is critical [10].
5.2 The Exercise “Vaccine”
Exercise offers a paradox: acute, heavy exertion can temporarily raise inflammatory markers, but chronic training lowers them [11]. Skeletal muscle acts as an endocrine organ. When muscles contract, they release myokines (muscle-derived IL-6) that paradoxically act as anti-inflammatory signals, blocking the pathways that lead to chronic inflammation.
5.3 Lifestyle Hygiene
- Oral Health: Periodontitis is a vascular threat. It creates a chronic bacterial load that enters the bloodstream. Treating gum disease has been shown to lower systemic CRP [12].
- Sleep: Both sleep deprivation and sleep apnea drive inflammation via oxidative stress. Correcting apnea (CPAP) and ensuring 7-8 hours of sleep are fundamental anti-inflammatory interventions [13, 14].
5.4 Pharmacotherapy
When lifestyle isn’t enough, we have tools:
- Statins: They are dual inhibitors. They lower cholesterol, but they also dampen the inflammatory response in the vessel wall [4].
- Colchicine: This is the game-changer for residual inflammatory risk. The COLCOT [15] and LoDoCo2 [16] trials showed that low-dose colchicine (0.5mg) significantly reduced cardiovascular events. It works by blocking the assembly of the NLRP3 inflammasome, stopping inflammation at the source.
- The Future: Drugs like Ziltivekimab (an IL-6 inhibitor) are currently in trials for patients with chronic kidney disease and high CRP, offering hope for even more targeted therapy.
VI. Conclusion
The debate is settled: Inflammation is a mechanism of disease, not just a bystander. The data clearly shows that hs-CRP belongs alongside ApoB and Lp(a) in a modern risk assessment.
We must move beyond the “plumbing” model. The goal of therapy is no longer just to lower a number on a lipid panel; it is to stabilize the biology of the artery. This requires a comprehensive strategy that lowers the particle burden to physiological levels while simultaneously extinguishing the inflammatory fire.
 References
References
- Ridker PM, Moorthy MV, Cook NR, Rifai N, Lee IM, Buring JE. Inflammation, Cholesterol, Lipoprotein(a), and 30-Year Cardiovascular Outcomes in Women. N Engl J Med. 2024;391(22):2087-2097. doi:10.1056/NEJMoa2405182
- Mensah GA, Arnold N, Prabhu SD, Ridker PM, Welty FK. Inflammation and Cardiovascular Disease: 2025 ACC Scientific Statement: A Report of the American College of Cardiology. J Am Coll Cardiol. Published online September 29, 2025. doi:10.1016/j.jacc.2025.08.047
- Libby P. The changing landscape of atherosclerosis. Nature. 2021;592(7855):524-533. doi:10.1038/s41586-021-03392-8
- Ridker PM, Cannon CP, Morrow D, et al. C-reactive protein levels and outcomes after statin therapy. N Engl J Med. 2005;352(1):20-28. doi:10.1056/NEJMoa042378
- Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359(21):2195-2207. doi:10.1056/NEJMoa0807646
- Sniderman AD, Williams K, Contois JH, et al. A meta-analysis of low-density lipoprotein cholesterol, non-high-density lipoprotein cholesterol, and apolipoprotein B as markers of cardiovascular risk. Circ Cardiovasc Qual Outcomes. 2011;4(3):337-345. doi:10.1161/CIRCOUTCOMES.110.959247
- McQueen MJ, Hawken S, Wang X, et al. Lipids, lipoproteins, and apolipoproteins as risk markers of myocardial infarction in 52 countries (the INTERHEART study): a case-control study. Lancet. 2008;372(9634):224-233. doi:10.1016/S0140-6736(08)61076-4
- Tsimikas S. A Test in Context: Lipoprotein(a): Diagnosis, Prognosis, Controversies, and Emerging Therapies. J Am Coll Cardiol. 2017;69(6):692-711. doi:10.1016/j.jacc.2016.11.042
- Estruch R, Ros E, Salas-Salvadó J, et al. Primary Prevention of Cardiovascular Disease with a Mediterranean Diet Supplemented with Extra-Virgin Olive Oil or Nuts. N Engl J Med. 2018;378(25):e34. doi:10.1056/NEJMoa1800389
- Sacks FM, Bray GA, Carey VJ, et al. Comparison of weight-loss diets with different compositions of fat, protein, and carbohydrates. N Engl J Med. 2009;360(9):859-873. doi:10.1056/NEJMoa0804748
- Fedewa MV, Hathaway ED, Ward-Ritacco CL. Effect of exercise training on C reactive protein: a systematic review and meta-analysis of randomised and non-randomised controlled trials. Br J Sports Med. 2017;51(8):670-676. doi:10.1136/bjsports-2016-095999
- Demmer RT, Trinquart L, Zuk A, et al. The influence of anti-infective periodontal treatment on C-reactive protein: a systematic review and meta-analysis of randomized controlled trials. PLoS One. 2013;8(10):e77441. Published 2013 Oct 14. doi:10.1371/journal.pone.0077441
- Meier-Ewert HK, Ridker PM, Rifai N, et al. Effect of sleep loss on C-reactive protein, an inflammatory marker of cardiovascular risk. J Am Coll Cardiol. 2004;43(4):678-683. doi:10.1016/j.jacc.2003.07.050
- Guo Y, Pan L, Ren D, Xie X. Impact of continuous positive airway pressure on C-reactive protein in patients with obstructive sleep apnea: a meta-analysis. Sleep Breath. 2013;17(2):495-503. doi:10.1007/s11325-012-0722-2
- Tardif JC, Kouz S, Waters DD, et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N Engl J Med. 2019;381(26):2497-2505. doi:10.1056/NEJMoa1912388
- Nidorf SM, Fiolet ATL, Mosterd A, et al. Colchicine in Patients with Chronic Coronary Disease. N Engl J Med. 2020;383(19):1838-1847. doi:10.1056/NEJMoa2021372