
The Genetic Architecture of Cardiovascular Protection: Validated Targets, Cumulative Exposure, and the Biology of Lifelong Risk
Cardiovascular disease remains the leading cause of morbidity and mortality globally, arising from a complex interplay of environmental exposures, metabolic dysregulation, vascular injury, thrombosis, and inherited susceptibility [1]. Despite major advances in prevention and treatment, atherosclerotic cardiovascular disease (ASCVD) continues to account for a disproportionate share of premature death and disability. Traditional risk factors—elevated low-density lipoprotein cholesterol (LDL-C), hypertension, diabetes mellitus, smoking, obesity, and sedentary behavior—remain central determinants of disease burden. Yet the modern understanding of atherogenesis has been fundamentally reshaped by human genetics [2], [3]. Over the past two decades, the field has moved well beyond family studies and observational epidemiology into the era of genome-wide association studies (GWAS), exome sequencing, large-scale biobanks, and Mendelian randomization. Together, these approaches have changed not only how atherosclerosis is studied, but how causality is assigned and how therapeutic targets are prioritized [3], [4].
One of the most consequential lessons from this genomic era is that naturally occurring cardioprotective variants are disproportionately informative. These variants, often rare loss-of-function alleles or function-reducing missense substitutions, act as experiments of nature. By partially or completely reducing the activity of a protein from birth, they approximate lifelong target inhibition and thereby provide unusually strong evidence about causality, magnitude of benefit, and long-term tolerability in humans [5], [6]. Human genetics has shown that persistent lowering of apoB-containing lipoproteins, acceleration of triglyceride-rich lipoprotein clearance, reduction of lipoprotein(a) burden, and attenuation of inflammatory signaling can each reduce the risk of coronary artery disease, myocardial infarction, ischemic stroke, and related vascular events [4], [7], [13], [17], [39], [40].
The importance of these variants is twofold. First, they provide causal evidence linking specific pathways to atherosclerosis, often more convincingly than conventional epidemiology. Because genetic variants are randomly allocated at conception and fixed across the lifespan, they are less vulnerable to reverse causation and many traditional forms of confounding [8], [35]. Second, they de-risk drug development. If lifelong partial inhibition of a protein lowers vascular risk without major human toxicity, that protein becomes an unusually attractive therapeutic target. PCSK9 is the clearest example of this model, but it is no longer unique. APOC3, ANGPTL3, NPC1L1, LPA, and IL6R have each moved from human genetic insight toward pharmacologic development, therapeutic validation, or both [5], [6], [16], [17], [21], [26], [39], [40].
At the same time, the evidence base is not uniform across all proposed cardioprotective genes. Some loci have direct human outcome-level validation and a clear mechanistic pathway to intervention. Others show convincing biomarker effects but less mature cardiovascular outcome evidence. Still others emerge from founder populations, exome scans, or pathway-based inference without yet yielding a fully resolved, therapeutically validated mechanism [4], [11]. For that reason, a rigorous evidence hierarchy is essential. Lower triglycerides, higher HDL-C, or lower LDL-C in variant carriers are not sufficient by themselves. The most clinically informative cardioprotective targets are those supported by convergent evidence linking genotype to mechanism, mechanism to biomarker, and biomarker to hard cardiovascular outcomes.
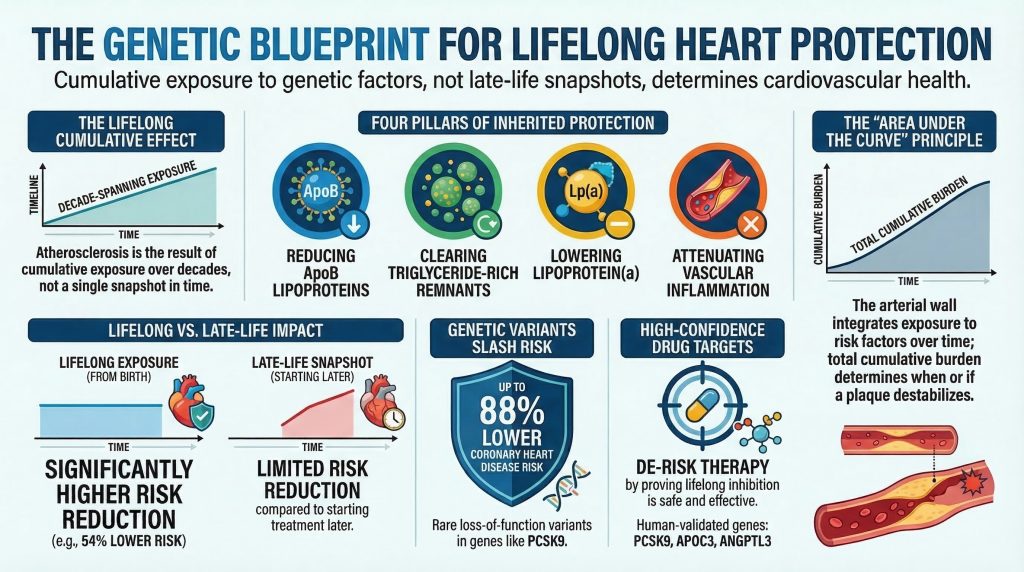
This review provides an evidence-graded analysis of the genetic architecture of cardiovascular protection. It first maps the major mechanistic domains through which inherited protection operates. It then examines the highest-confidence cardioprotective pathways and genes—those supported by human outcome evidence and, in several cases, therapeutic translation—including PCSK9, APOC3, ANGPTL3, LPL, NPC1L1, LPA, and IL6R. It next evaluates biologically compelling but less fully validated targets, including ANGPTL4, ASGR1, CETP, HMGCR, favorable LDLR and APOB variation, SORT1, and emerging exome signals such as CIDEB and ZNRF3 [4], [9], [11], [31], [38]. It also considers the special role of founder populations as discovery platforms, while distinguishing hypothesis-generating isolates from established cardioprotective mechanisms. Finally, it argues that the most important lesson from human genetics may be conceptual rather than taxonomic: lifetime vascular risk is better explained by cumulative inherited exposure than by any single clinical snapshot obtained later in life [8], [12], [34].
Mechanistic Pathway Map: Four Pillars of Inherited Cardiovascular Protection
The architecture of inherited cardioprotection is most usefully understood not as a list of isolated genes, but as a set of recurrent mechanistic domains. Current evidence suggests that robust genetic protection against ASCVD clusters within four principal axes: (1) lifelong reduction in apoB-containing lipoprotein burden, (2) enhanced hydrolysis and clearance of triglyceride-rich lipoproteins and their remnants, (3) reduction in lipoprotein(a)-mediated atherothrombotic burden, and (4) attenuation of vascular inflammation [3], [7], [13], [15], [17], [39].
The first pillar is lifelong reduction of apoB-containing lipoproteins, especially LDL particles. ApoB-containing particles are the necessary causal particles in atherosclerosis. Their entry into and retention within the arterial intima initiates the cascade of oxidation, endothelial activation, monocyte recruitment, foam-cell formation, and plaque progression that ultimately underlies most myocardial infarction and ischemic stroke [13]. Genes operating in this domain include PCSK9, NPC1L1, LDLR, APOB, HMGCR, and, in a more regulatory sense, SORT1 [6], [9], [13], [38]. Protective variants in these genes lower the concentration of atherogenic particles or reduce cumulative lifetime exposure to them, thereby decreasing the probability that plaque will form, enlarge, and destabilize.
The second pillar is the metabolism of triglyceride-rich lipoproteins (TRLs), including very-low-density lipoproteins (VLDL), chylomicrons, and their cholesterol-rich remnants. Although LDL remains the canonical atherogenic particle, human genetic evidence now strongly supports a causal role for TRL remnants in ASCVD [7], [15]. APOC3, ANGPTL3, ANGPTL4, and LPL are key regulators of this domain. These genes alter the hydrolysis and clearance of TRLs by modulating lipoprotein lipase activity, endothelial lipase activity, and hepatic remnant uptake [15], [16], [22], [27]. Variants that enhance TRL catabolism lower remnant cholesterol and reduce vascular risk.
A third pillar, often underemphasized in older lipid-centric models, is lipoprotein(a), or Lp(a). Lp(a) is an apoB-containing particle covalently linked to apolipoprotein(a), whose size and concentration are largely determined by genetic variation at the LPA locus [39], [40]. Unlike LDL alone, Lp(a) appears to couple atherogenic cholesterol delivery with proinflammatory and prothrombotic properties. Human genetics has provided some of the strongest causal evidence in cardiovascular biology that elevated Lp(a) increases coronary risk, while genetically lower Lp(a) confers protection [39], [40].
The fourth pillar is inflammation. Atherosclerosis is not merely a lipid storage disorder, but a chronic inflammatory disease of the arterial wall. Innate and adaptive immune signaling influence plaque initiation, growth, necrotic core formation, fibrous cap stability, and thrombogenicity. The clearest genetically supported anti-inflammatory cardioprotective pathway is IL-6 signaling, particularly through functional variation in IL6R [17]. Although this pathway does not yet have the same degree of pharmacologic cardiovascular outcome validation as PCSK9, the combination of human genetic evidence and anti-inflammatory trial data has firmly established inflammation as a causal domain in ASCVD [17], [18].
These four pillars are interdependent rather than autonomous. ApoB particles provoke vascular inflammation. Remnants contribute to endothelial dysfunction and foam-cell formation. Lp(a) links lipid biology to thrombosis and inflammation. Inflammatory cytokines influence hepatic lipoprotein production, endothelial activation, and plaque instability. Even so, this framework provides a useful map for evaluating cardioprotective targets and for understanding why certain genes repeatedly emerge as human-validated protective mechanisms.
From Locus Discovery to Causal Biology: Why Genetics Matters
The strength of human genetics lies not merely in its ability to detect association, but in its capacity to rank causal mechanisms. Mendelian disease studies first demonstrated that rare, high-effect mutations could reveal broadly relevant biology. Familial hypercholesterolemia established the centrality of LDL receptor biology; rare PCSK9 mutations later showed that disruption of the same pathway could be either harmful or protective, depending on directionality [2], [5], [20]. Similarly, rare Mendelian dyslipidemias anticipated later population-level findings showing that common and rare variants often converge on the same pathways [2].
GWAS added another dimension by revealing that common variants of small effect could still identify therapeutically decisive biology. Indeed, one of the most important lessons of cardiovascular genetics is that the variance explained by a variant is not the same as the biological or therapeutic importance of its gene [2], [3]. Common variants near HMGCR or NPC1L1 have small effects on LDL-C, but the proteins they implicate became targets of effective therapies [6], [9], [14], [30]. Conversely, some large-effect loci have remained mechanistically opaque for years, particularly when the causal signal resides in non-coding regulatory sequence rather than protein-coding variation [2], [3].
A canonical example of successful movement from association to mechanism is the chromosome 1p13 locus near SORT1. Initial GWAS linked the region to LDL-C and myocardial infarction risk, but the biology was uncertain. Subsequent functional work identified regulatory variants that altered SORT1 expression in the liver, thereby affecting lipoprotein secretion and LDL metabolism [38]. This case established that non-coding association signals can indeed be translated into specific genes and specific mechanisms, though such successes remain the exception rather than the rule. By contrast, loci such as 9p21 illustrate that a strong and reproducible association with coronary artery disease does not necessarily yield rapid mechanistic clarity [2], [3].
For cardioprotective genetics, then, the highest-confidence targets tend to be those where association, coding perturbation, biologic mechanism, biomarker shift, and outcome reduction all align. That is the standard used below.
Tier 1: High-Confidence Cardioprotective Targets with Human Outcome Support
The most rigorous definition of a cardioprotective gene requires more than a favorable biomarker profile. It requires human evidence that function-altering variants in the gene are associated with lower incidence of cardiovascular events, ideally reinforced by mechanistic plausibility and therapeutic translation. On that standard, a limited number of pathways stand out.
PCSK9 (Proprotein Convertase Subtilisin/Kexin Type 9)
PCSK9 is the archetypal success story of cardiovascular genetics and remains the clearest demonstration of the path from human loss-of-function genetics to precision therapy. PCSK9 is synthesized predominantly in the liver as a secreted serine protease that regulates the abundance of the LDL receptor (LDLR) on hepatocytes [19]. Its essential action is to bind LDLR and redirect it toward lysosomal degradation rather than recycling. Under normal conditions, LDLR internalizes circulating LDL particles, releases cargo in the acidic endosomal compartment, and then returns to the cell surface. PCSK9 disrupts this recycling process and therefore reduces hepatocyte LDLR density [19], [20].
The importance of PCSK9 first became evident when gain-of-function mutations were identified as a cause of autosomal dominant hypercholesterolemia [20]. The field changed when naturally occurring loss-of-function variants, including Y142X, C679X, and R46L, were identified in population cohorts [5]. Carriers had increased hepatic LDL receptor density, enhanced LDL clearance, and lifelong lower LDL-C levels. In the landmark study by Cohen and colleagues, Black carriers of nonsense variants in PCSK9 had an approximately 28% reduction in LDL-C and an 88% lower risk of coronary heart disease, while White carriers of R46L had more modest LDL-C lowering but still significant protection [5]. The finding was transformative because it showed that lifelong LDL lowering from birth produces much larger proportional risk reductions than short-term treatment initiated later in life.
The translational consequences were immediate. Monoclonal antibodies such as evolocumab and alirocumab, and later silencing approaches such as inclisiran, were developed to mimic the protective genetic phenotype. Outcome trials demonstrated that pharmacologic PCSK9 inhibition markedly lowers LDL-C on top of statins and reduces major cardiovascular events [21]. PCSK9 is therefore not just a well-supported target; it is the paradigm by which contemporary cardioprotective genetics is judged.
NPC1L1 (Niemann-Pick C1-Like 1)
NPC1L1 is the principal transporter mediating intestinal cholesterol absorption. It is expressed in the brush border of enterocytes and facilitates uptake of both dietary and biliary sterols [6]. Rare inactivating mutations in NPC1L1 were identified in large exome sequencing datasets and were associated with modest lifelong reductions in LDL-C but disproportionately large reductions in coronary heart disease risk [6]. In the study by Stitziel and colleagues, carriers had approximately 12 mg/dL lower LDL-C and about 53% lower risk of coronary disease [6]. Although the magnitude of effect in rare variant carriers should not be extrapolated uncritically to general-population treatment effects, the result strongly reinforced a key principle of cardiovascular biology: modest LDL differences, when sustained over decades, can yield very large cumulative risk differences.
NPC1L1 also exemplifies the convergence of genetics and therapeutics. Ezetimibe inhibits NPC1L1 pharmacologically, and the IMPROVE-IT trial showed that adding ezetimibe to statin therapy lowers cardiovascular events [30]. Thus, as with PCSK9, human genetics anticipated clinical benefit and strengthened causal confidence in the target.
APOC3 (Apolipoprotein C-III)
APOC3 is one of the major human regulators of triglyceride-rich lipoprotein metabolism. Synthesized largely by the liver and intestine, apolipoprotein C-III resides on the surface of chylomicrons, VLDL, and HDL particles, where it inhibits lipoprotein lipase, delays lipolysis of triglyceride-rich particles, and impairs hepatic remnant uptake [15], [22]. Elevated APOC3 therefore promotes accumulation of triglycerides and remnant cholesterol, both increasingly recognized as causal mediators of ASCVD.
Exome sequencing studies identified rare loss-of-function mutations in APOC3, including R19X and splice-disrupting alleles, that lower APOC3 levels and reduce plasma triglycerides by roughly 40% [23], [24]. More importantly, these variants were associated with lower risk of ischemic vascular disease and coronary disease [23], [24]. These observations strengthened the case that triglyceride-rich remnants are not merely correlated with risk, but are themselves causally involved in atherosclerosis. The likely benefit appears to be mediated primarily through improved remnant clearance rather than major LDL reduction.
The APOC3 story has also translated rapidly. Antisense oligonucleotide and RNA-targeted approaches that suppress APOC3 have shown marked triglyceride lowering in severe hypertriglyceridemia. Cardiovascular outcomes data are still maturing, but genetically APOC3 already belongs among the strongest validated targets for residual risk beyond LDL.
ANGPTL3 (Angiopoietin-Like 3)
ANGPTL3 is a liver-derived secretory protein that inhibits both lipoprotein lipase and endothelial lipase, placing it at a central junction in systemic lipid trafficking [16], [25]. Homozygous loss-of-function mutations in ANGPTL3 cause familial combined hypolipidemia, characterized by low LDL-C, low triglycerides, and low HDL-C [25]. This phenotype is notable because it simultaneously alters several lipid compartments relevant to vascular disease.
Population-based sequencing studies showed that heterozygous carriers of inactivating ANGPTL3 variants have lower plasma lipid levels and approximately 34% lower risk of coronary artery disease [16]. The protective signature is broader than in a purely LDL-centered pathway: lower triglycerides, lower remnant cholesterol, lower apoB, and lower LDL-C all contribute. Some LDL lowering appears partly independent of canonical LDL receptor biology, increasing the therapeutic interest of ANGPTL3 in disorders such as homozygous familial hypercholesterolemia [16], [25].
This promise has been realized in part through evinacumab, a monoclonal antibody against ANGPTL3, which lowers LDL-C substantially even in settings where LDLR-dependent therapies are limited [26]. ANGPTL3 therefore stands with PCSK9 and NPC1L1 as one of the clearest examples of protective human genetics guiding successful translational development.
LPL (Lipoprotein Lipase)
Lipoprotein lipase is the key effector enzyme for hydrolysis of triglycerides within chylomicrons and VLDL, thereby facilitating tissue fatty acid uptake and remnant clearance [15], [28]. If APOC3 and ANGPTL proteins act as brakes on triglyceride clearance, LPL is the engine that confers protection when activated or disinhibited.
The best-known naturally occurring protective variant in LPL is S447X (Ser447Ter, S447*), a near-terminal truncating variant associated with enhanced lipolytic efficiency, lower triglycerides, higher HDL-C, and lower coronary risk [29]. Although the exact biochemical mechanism has been debated, the epidemiologic signal has been consistent. More broadly, coding variation in and around the LPL pathway has repeatedly shown that greater lipolytic clearance of TRLs and remnants is cardioprotective [4].
LPL is conceptually important because it provides reciprocal validation of the remnant hypothesis. If inhibiting APOC3 or ANGPTL proteins lowers cardiovascular risk by releasing the LPL brake, then enhancing LPL itself should be protective. Human genetics confirms exactly that.
LPA and Lipoprotein(a)
Any contemporary discussion of inherited cardioprotection is incomplete without lipoprotein(a). Lp(a) levels are determined predominantly by variation at the LPA locus, particularly by apo(a) isoform size and related genetic structure [39], [40]. Elevated Lp(a) is now among the most strongly genetically supported causal risk factors for coronary disease. Conversely, inherited reductions in Lp(a) confer protection [39], [40].
The key conceptual importance of LPA is that it broadens the field beyond standard LDL biology. Lp(a) appears to promote atherosclerosis through apoB-mediated arterial retention while also exerting proinflammatory and prothrombotic effects. Genetic studies have shown that variants associated with elevated Lp(a) increase myocardial infarction risk, while Mendelian-randomization analyses support causality rather than mere correlation [39], [40]. Although naturally occurring protective LPA variants are less neatly framed as classic loss-of-function alleles than PCSK9 or APOC3, the pathway is sufficiently validated to merit inclusion among high-confidence cardioprotective mechanisms. Therapeutic programs targeting Lp(a) now represent one of the most promising frontiers in preventive cardiology.
IL6R and IL-6 Signaling
IL6R occupies a special place in the cardioprotective landscape because it extends validated protection beyond lipoprotein metabolism. Atherosclerosis is a chronic inflammatory disease, and IL-6 is a central mediator of acute-phase and vascular inflammatory signaling [17], [18]. The common Asp358Ala variant in IL6R increases receptor shedding and attenuates classical IL-6 signaling. Carriers have lower CRP and fibrinogen levels and lower coronary heart disease risk [17].
The magnitude of vascular protection is smaller than that seen with some lipid pathways, and IL6R has not yet reached the same level of definitive pharmacologic cardiovascular outcome validation as PCSK9. Nevertheless, the mechanistic significance is profound. It shows that genetically reduced inflammatory signaling can reduce ASCVD risk independently of lipid levels. This genetic result complements interventional evidence from anti-inflammatory trials such as CANTOS, even though CANTOS targeted IL-1β rather than IL-6 directly [18]. IL6R is therefore best viewed as a genetically validated protective pathway with strong translational promise.
Tier 2: Biologically Compelling but Less Fully Validated Targets
A second tier includes genes and pathways with substantial biologic credibility and often convincing biomarker effects, but with less complete human outcome evidence, less certain mechanism, or less mature therapeutic translation.
ANGPTL4
ANGPTL4, like ANGPTL3, inhibits lipoprotein lipase and influences fasting-state lipid partitioning [27]. The E40K missense variant reduces inhibitory activity and is associated with lower triglycerides, higher HDL-C, and reduced coronary disease risk [4], [28]. These observations strongly support the general principle that LPL disinhibition is cardioprotective. However, ANGPTL4 does not yet have the same breadth of outcome validation or therapeutic maturity as PCSK9, ANGPTL3, or APOC3. In addition, animal studies raised safety concerns with more complete pathway disruption under certain dietary conditions [27]. Human genetics supports ANGPTL4 as an important pathway-supported target, but not yet as a top-tier clinically validated one.
ASGR1
ASGR1 is a hepatic receptor involved in clearance of desialylated glycoproteins. Rare loss-of-function variants were associated with lower non-HDL cholesterol and reduced coronary artery disease risk in Icelandic sequencing studies [31]. The signal is intriguing and potentially important, but the mechanism remains incompletely resolved and therapeutic translation is still early. ASGR1 is therefore best placed among promising cardioprotective loci rather than among the most validated targets.
CETP
CETP facilitates exchange of cholesteryl esters and triglycerides between HDL and apoB-containing lipoproteins. Variants that reduce CETP activity substantially raise HDL-C and may modestly lower apoB and LDL-C [9], [32]. Human genetics and the long arc of CETP inhibitor development have clarified an important point: whatever cardiovascular benefit CETP inhibition may confer is likely mediated by lowering apoB-containing particles, not by raising HDL per se [9], [32]. That distinction is crucial, because CETP was long misinterpreted as an HDL story when it is more properly an apoB story. Clinical trial results have been heterogeneous, reflecting both off-target toxicity and varying efficacy across compounds. CETP therefore remains pathway-supported but not definitively validated at the level of the strongest cardioprotective genes.
HMGCR, LDLR, and APOB Favorable Variation
These genes reinforce the centrality of cumulative LDL exposure. HMGCR variation has been especially informative because common variants such as rs12916 genetically mimic partial statin exposure over the lifespan [9]. Carriers have lower LDL-C and lower coronary risk, but also slightly increased diabetes risk, mirroring a known tradeoff of statin therapy. This is a powerful example of how genetics can predict both therapeutic benefit and mechanism-based adverse effects. Favorable regulatory variation in LDLR and APOB likewise supports the centrality of lifelong apoB lowering, even though the most dramatic pathogenic consequences of these genes are seen when function is impaired in the opposite direction [13], [14].
SORT1 and the 1p13 Locus
SORT1 merits discussion because it represents one of the best examples of successful translation from GWAS signal to specific mechanism. Variants at 1p13 are associated with LDL-C and myocardial infarction risk; functional studies showed that regulatory changes at the locus alter hepatic SORT1 expression and lipoprotein metabolism [38]. The locus is therefore highly informative biologically, but its role is more as a mechanistically illuminating regulatory node than as a classical human loss-of-function protective target with direct therapeutic analogy.
CIDEB, ZNRF3, SVEP1, KLF14, and Emerging Exome Signals
Large-scale sequencing continues to identify loci that may inform future therapeutic development. CIDEB appears relevant to hepatic lipid droplet biology and may influence both liver disease and cardiometabolic phenotypes [11]. ZNRF3 has emerged as a possible LDL-lowering target without obvious adverse glycemic or hepatic signatures, though the evidence remains early [11]. SVEP1 is notable because coding variation has been associated with coronary risk [4], but its directionality implicates vascular biology more than protective loss-of-function. KLF14 is metabolically interesting, especially in adipose biology, yet remains less directly validated as a cardiovascular protection target. These loci are best viewed as discovery-stage or pathway-informative rather than therapeutically settled.
Evidence Grading: A Practical Hierarchy of Cardioprotective Targets
A useful synthesis integrates four dimensions: (1) nature of genetic perturbation, (2) biomarker effect, (3) cardiovascular outcome evidence, and (4) degree of therapeutic translation.
Tier 1: highest-confidence cardioprotective targets
- PCSK9: major lifelong LDL lowering, strong human outcome evidence, approved therapies [5], [21]
- NPC1L1: modest lifelong LDL lowering with large rare-carrier outcome effects, approved therapy [6], [30]
- APOC3: major triglyceride/remnant lowering with strong human outcome support, advanced targeted therapies [23], [24]
- ANGPTL3: broad lipid lowering with human outcome support and approved therapy [16], [26]
- LPL: direct effector of remnant clearance with consistent human protective evidence [4], [29]
- LPA pathway: strong causal evidence that genetically lower Lp(a) reduces ASCVD risk, active therapeutic translation [39], [40]
- IL6R pathway: strong human genetic support for inflammatory causality, translational development underway [17], [18]
Tier 2: biologically compelling, but less fully validated
- ANGPTL4 [4], [27], [28]
- ASGR1 [31]
- CETP [9], [32]
- HMGCR [9], [14]
- Favorable LDLR/APOB variation [13], [14]
- SORT1 [38]
- CIDEB, ZNRF3, KLF14, and related emerging exome signals [11]
This hierarchy emphasizes a central principle: the most useful cardioprotective genes are not necessarily those with the largest biomarker effects, but those for which genetics, mechanism, outcomes, and therapeutic tractability all align.
Founder Populations and Isolates: Discovery Platforms, Not Evidence Substitutes
Founder populations occupy a privileged place in human genetics because isolation, bottlenecks, and endogamy can amplify otherwise rare variants, thereby increasing power to detect disease-modifying alleles. This logic has already yielded important discoveries in cardiometabolic biology, including protective lipid variants in relatively isolated populations [23], [24]. Such populations are especially valuable for hypothesis generation and rare variant discovery.
However, founder status is not itself evidence of cardioprotection. Claims regarding unusual vascular protection in specific isolates should be evaluated with the same rigor as any other proposed mechanism: the variant must be identified, its functional direction demonstrated, and its relationship to hard cardiovascular outcomes established. In the absence of that sequence of evidence, such populations remain discovery opportunities rather than validated examples of inherited protection. This distinction is particularly important when media narratives outpace peer-reviewed mechanistic literature.
Why Inherited Exposure Outperforms Late-Life Snapshots
The most important conceptual lesson from cardioprotective genetics is that atherosclerosis is fundamentally a disease of cumulative exposure. Clinical risk assessment traditionally relies on measurements taken in midlife: an LDL-C value, a blood pressure reading, an HbA1c, a high-sensitivity CRP. These are clinically useful, but biologically they are snapshots [12]. The arterial wall, by contrast, integrates exposure over decades. What matters mechanistically is the area under the curve of apoB burden, remnant cholesterol, Lp(a), blood pressure, glycemic injury, and inflammatory signaling [8], [13], [34].
Mendelian-randomization studies have shown that lifelong genetically mediated LDL lowering produces much larger reductions in coronary risk than the same absolute LDL reduction achieved pharmacologically for only a few years in adulthood [8]. Ference and colleagues demonstrated that a 1 mmol/L lower LDL-C level sustained from early life is associated with an approximately 54% lower risk of coronary heart disease [8]. This is far greater than the relative risk reduction usually observed when the same LDL difference is achieved later in life with treatment [8], [14]. The explanation is straightforward: lifelong protection prevents plaque from forming in the first place, whereas later therapy often modifies an already established disease process.
The same principle almost certainly extends beyond LDL. Persistently lower remnant burden, lower Lp(a), lower blood pressure, and lower inflammatory signaling from early life should all be expected to produce larger lifetime benefits than late correction alone. Human genetics is uniquely valuable here because it measures stable inherited predisposition rather than transient physiology. Genetic variants are fixed at conception, unaffected by most short-term environmental noise, and less prone to reverse causality [35]. In this sense, genetics allows researchers to look through day-to-day biologic fluctuation and infer a person’s long-term exposure architecture.
Polygenic Risk and the Continuum Between Monogenic and Common Variation
Rare monogenic protective variants are highly informative but uncommon. At the population level, inherited risk and protection are often polygenic. Polygenic risk scores aggregate the effects of many common variants across the genome to estimate baseline susceptibility to coronary disease [10], [36], [37]. These tools have shown that a subset of individuals carry polygenic risk approaching that seen in some monogenic disorders, particularly when interpreted together with conventional clinical information [36], [37].
The key contribution of polygenic models is not that they replace causal pathway biology, but that they complement it. Rare variants reveal mechanisms. Polygenic scores capture background burden. Together they suggest that inherited cardiovascular protection exists on a continuum: from rare, high-impact perturbations such as PCSK9 or APOC3 loss-of-function, to common regulatory variation in pathways such as HMGCR or CETP, to broad genome-wide architectures that influence lifelong arterial injury in aggregate.
PRS still requires caution. Performance varies across ancestry groups, calibration remains an active area of research, and most claims about event prevention remain inferential rather than trial-proven [36], [37]. Still, the broader message is clear: inherited biology can identify risk and protection much earlier than clinical disease becomes apparent.
Conclusion
Human genetics has fundamentally altered both the biology and strategy of cardiovascular prevention. It has shown that inherited cardiovascular protection is not diffuse or random, but concentrated in a limited number of causal domains: lifelong lowering of apoB-containing lipoproteins, more efficient clearance of triglyceride-rich remnants, reduction of Lp(a)-mediated atherothrombotic burden, and attenuation of vascular inflammation. Within these domains, a relatively small set of genes and pathways—especially PCSK9, NPC1L1, APOC3, ANGPTL3, LPL, LPA, and IL6R—now carry the strongest evidence as high-confidence cardioprotective mechanisms [5], [6], [16], [17], [21], [23], [24], [39], [40].
These pathways have done more than explain disease. They have provided a roadmap for therapy. PCSK9 inhibition is now standard clinical practice for selected patients. NPC1L1 genetics anticipated the benefit of ezetimibe. ANGPTL3 inhibition is clinically useful in severe dyslipidemia. APOC3 and Lp(a)-directed strategies continue to extend the frontier of precision prevention. IL6R has strengthened the case that inflammatory signaling is not merely associated with atherosclerosis, but causally involved in it.
Yet the most important lesson may be even broader. Cardiovascular risk is best understood not as a static trait measured in middle age, but as the cumulative consequence of decades of exposure. Human genetics provides a uniquely powerful lens on that cumulative process. Whether through rare cardioprotective alleles, regulatory variation in key pathways, or polygenic background burden, inherited variation reveals the tempo of arterial injury long before disease is clinically visible. The future of preventive cardiology will therefore depend not only on identifying more loci, but on learning to mimic nature’s most protective perturbations earlier, more precisely, and with greater mechanistic confidence.
References
-
- World Health Organization, “Cardiovascular diseases (CVDs),” WHO, 2025. https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds)
- Kathiresan S, Srivastava D. Genetics of human cardiovascular disease. Cell. 2012;148(6):1242-1257. doi:10.1016/j.cell.2012.03.001
- Khera AV, Kathiresan S. Genetics of coronary artery disease: discovery, biology and clinical translation. Nat Rev Genet. 2017;18(6):331-344. doi:10.1038/nrg.2016.160
- Myocardial Infarction Genetics and CARDIoGRAM Exome Consortia Investigators, Stitziel NO, Stirrups KE, et al. Coding Variation in ANGPTL4, LPL, and SVEP1 and the Risk of Coronary Disease. N Engl J Med. 2016;374(12):1134-1144. doi:10.1056/NEJMoa1507652
- Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354(12):1264-1272. doi:10.1056/NEJMoa054013.
- Myocardial Infarction Genetics Consortium Investigators, Stitziel NO, Won HH, et al. Inactivating mutations in NPC1L1 and protection from coronary heart disease.
- Budoff M. Triglycerides and Triglyceride-Rich Lipoproteins in the Causal Pathway of Cardiovascular Disease. Am J Cardiol. 2016;118(1):138-145. doi:10.1016/j.amjcard.2016.04.004
- Ference BA, Yoo W, Alesh I, et al. Effect of long-term exposure to lower low-density lipoprotein cholesterol beginning early in life on the risk of coronary heart disease: a Mendelian randomization analysis. J Am Coll Cardiol. 2012;60(25):2631-2639. doi:10.1016/j.jacc.2012.09.017.
- Ference BA, Kastelein JJP, Ginsberg HN, et al. Association of Genetic Variants Related to CETP Inhibitors and Statins With Lipoprotein Levels and Cardiovascular Risk. JAMA. 2017;318(10):947-956. doi:10.1001/jama.2017.11467.
- Inouye M, Abraham G, Nelson CP, et al. Genomic Risk Prediction of Coronary Artery Disease in 480,000 Adults: Implications for Primary Prevention. J Am Coll Cardiol. 2018;72(16):1883-1893. doi:10.1016/j.jacc.2018.07.079
- Nielsen JB, Rom O, Surakka I, et al. Loss-of-function genomic variants highlight potential therapeutic targets for cardiovascular disease. Nat Commun. 2020;11(1):6417. Published 2020 Dec 18. doi:10.1038/s41467-020-20086-3.
- Lloyd-Jones DM, Braun LT, Ndumele CE, et al. Use of Risk Assessment Tools to Guide Decision-Making in the Primary Prevention of Atherosclerotic Cardiovascular Disease: A Special Report From the American Heart Association and American College of Cardiology.
- Ference BA, Ginsberg HN, Graham I, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel.
- Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366(9493):1267-1278. doi:10.1016/S0140-6736(05)67394-1.
- Ginsberg HN, Packard CJ, Chapman MJ, et al. Triglyceride-rich lipoproteins and their remnants: metabolic insights, role in atherosclerotic cardiovascular disease, and emerging therapeutic strategies-a consensus statement from the European Atherosclerosis Society.
- Dewey FE, Gusarova V, Dunbar RL, et al. Genetic and Pharmacologic Inactivation of ANGPTL3 and Cardiovascular Disease. N Engl J Med. 2017;377(3):211-221. doi:10.1056/NEJMoa1612790.
- IL6R Genetics Consortium Emerging Risk Factors Collaboration, Sarwar N, Butterworth AS, et al. Interleukin-6 receptor pathways in coronary heart disease: a collaborative meta-analysis of 82 studies. Lancet. 2012;379(9822):1205-1213. doi:10.1016/S0140-6736(11)61931-4.
- Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease.
- Horton JD, Cohen JC, Hobbs HH. PCSK9: a convertase that coordinates LDL catabolism.
- Abifadel M, Varret M, Rabès JP, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia.
- Sabatine MS, Giugliano RP, Keech AC, et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease.
- Ramms B, Gordts PLSM. Apolipoprotein C-III in triglyceride-rich lipoprotein metabolism.
- Jørgensen AB, Frikke-Schmidt R, Nordestgaard BG, Tybjærg-Hansen A. Loss-of-function mutations in APOC3 and risk of ischemic vascular disease.
- TG and HDL Working Group of the Exome Sequencing Project, National Heart, Lung, and Blood Institute, Crosby J, Peloso GM, et al. Loss-of-function mutations in APOC3, triglycerides, and coronary disease.
- Musunuru K, Kathiresan S. Surprises From Genetic Analyses of Lipid Risk Factors for Atherosclerosis.
- Raal FJ, Rosenson RS, Reeskamp LF, et al. Evinacumab for Homozygous Familial Hypercholesterolemia.
- Aryal B, Price NL, Suarez Y, Fernández-Hernando C. ANGPTL4 in Metabolic and Cardiovascular Disease.
- Romeo S, Yin W, Kozlitina J, et al. Rare loss-of-function mutations in ANGPTL family members contribute to plasma triglyceride levels in humans.
- Sun W, Wu Y, Wen Y, Guo M, Zhang H. The association of the S447X mutation in LPL with Coronary artery disease: a meta-analysis. Minerva Cardioangiol. 2019;67(3):246-253. doi:10.23736/S0026-4725.18.04668-6.
- Cannon CP, Blazing MA, Giugliano RP, et al. Ezetimibe Added to Statin Therapy after Acute Coronary Syndromes.
- Nioi P, Sigurdsson A, Thorleifsson G, et al. Variant ASGR1 Associated with a Reduced Risk of Coronary Artery Disease.
- Nelson AJ, Sniderman AD, Ditmarsch M, et al. Cholesteryl Ester Transfer Protein Inhibition Reduces Major Adverse Cardiovascular Events by Lowering Apolipoprotein B Levels.
- Vassy JL, Christensen KD, Schonman EF, et al. The Impact of Whole-Genome Sequencing on the Primary Care and Outcomes of Healthy Adult Patients: A Pilot Randomized Trial.
- Ference BA, Braunwald E, Catapano AL. The LDL cumulative exposure hypothesis: evidence and practical applications.
- Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data.
- Khera AV, Chaffin M, Aragam KG, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet. 2018;50(9):1219-1224. doi:10.1038/s41588-018-0183-z
- Inouye M, Abraham G, Nelson CP, et al. Genomic Risk Prediction of Coronary Artery Disease in 480,000 Adults: Implications for Primary Prevention. J Am Coll Cardiol. 2018;72(16):1883-1893. doi:10.1016/j.jacc.2018.07.079
- Musunuru K, Strong A, Frank-Kamenetsky M, et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 2010;466(7307):714-719. doi:10.1038/nature09266
- Clarke R, Peden JF, Hopewell JC, et al. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med. 2009;361(26):2518-2528. doi:10.1056/NEJMoa0902604.
- Kamstrup PR, Tybjaerg-Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301(22):2331-2339. doi:10.1001/jama.2009.801