
The historical conceptualization of atherosclerosis has frequently focused on a linear relationship between circulating cholesterol levels and the development of obstructive coronary artery disease. However, contemporary genomic research, particularly large-scale genome-wide association studies and Mendelian randomization analyses, has unveiled a significantly more complex landscape [1]. It is now evident that certain individuals possess a “super-resilient” vascular phenotype, characterized by the ability to withstand high levels of systemic risk factors without developing rupture-prone atherosclerotic lesions [1]. This resilience is driven by a series of discrete genetic factors that modulate the vascular response at multiple levels: systemic and local inflammatory signaling, structural integrity of the vessel wall, transendothelial transport of lipoproteins, and the innate immunological neutralization of pro-oxidative threats [1]. Central to this understanding are the roles played by the interleukin-6 receptor variant p.Asp358Ala [1–3], the transport protein MIA3/TANGO1 [6,7], the scavenger receptor SCARB1 [8–10], the pioneer transcription factor PBX1 [11–13], and the protective activity of natural autoantibodies against oxidized phospholipids, such as E06 [14–16].
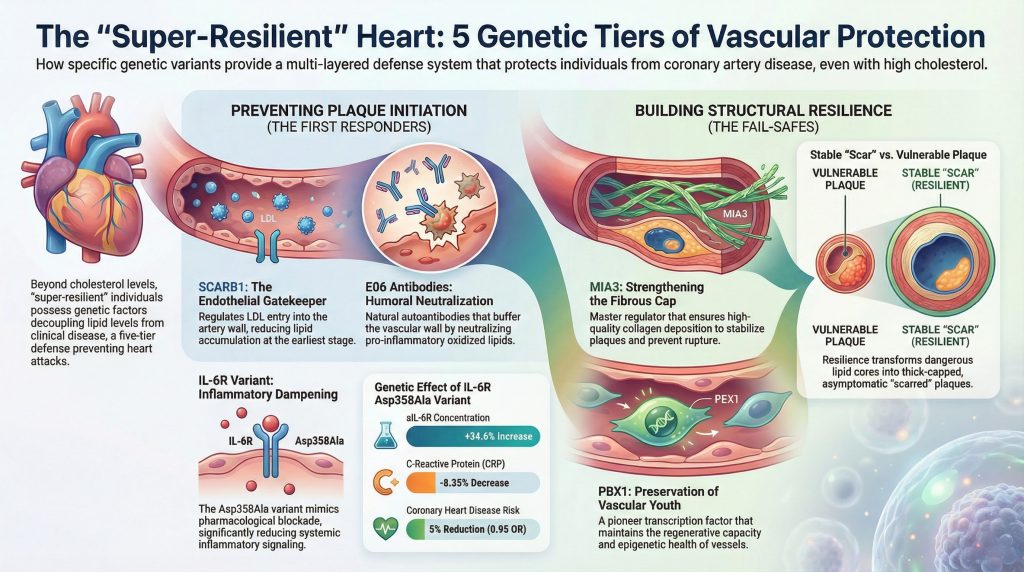
The Inflammatory Modulation of the IL-6 Receptor Pathway
Inflammation is a fundamental driver of atherogenesis, serving as the bridge between lipid deposition and clinical events such as myocardial infarction [1]. Among the cytokines implicated in this process, interleukin-6 (IL-6) occupies a central position as a pleiotropic regulator of the immune response, synthesized primarily by macrophages, T cells, and adipocytes [2]. The signaling architecture of IL-6 is bifurcated into classical signaling—occurring via the membrane-bound IL-6 receptor (mIL-6R)—and trans-signaling, mediated by the soluble form of the receptor (sIL-6R) [2]. While classical signaling is restricted to cells expressing mIL-6R (such as hepatocytes and certain immune subsets), trans-signaling allows IL-6 to activate any cell expressing the signal-transducing subunit gp130, thereby broadening the pro-inflammatory reach of the cytokine to the vascular endothelium and other critical tissues [2].
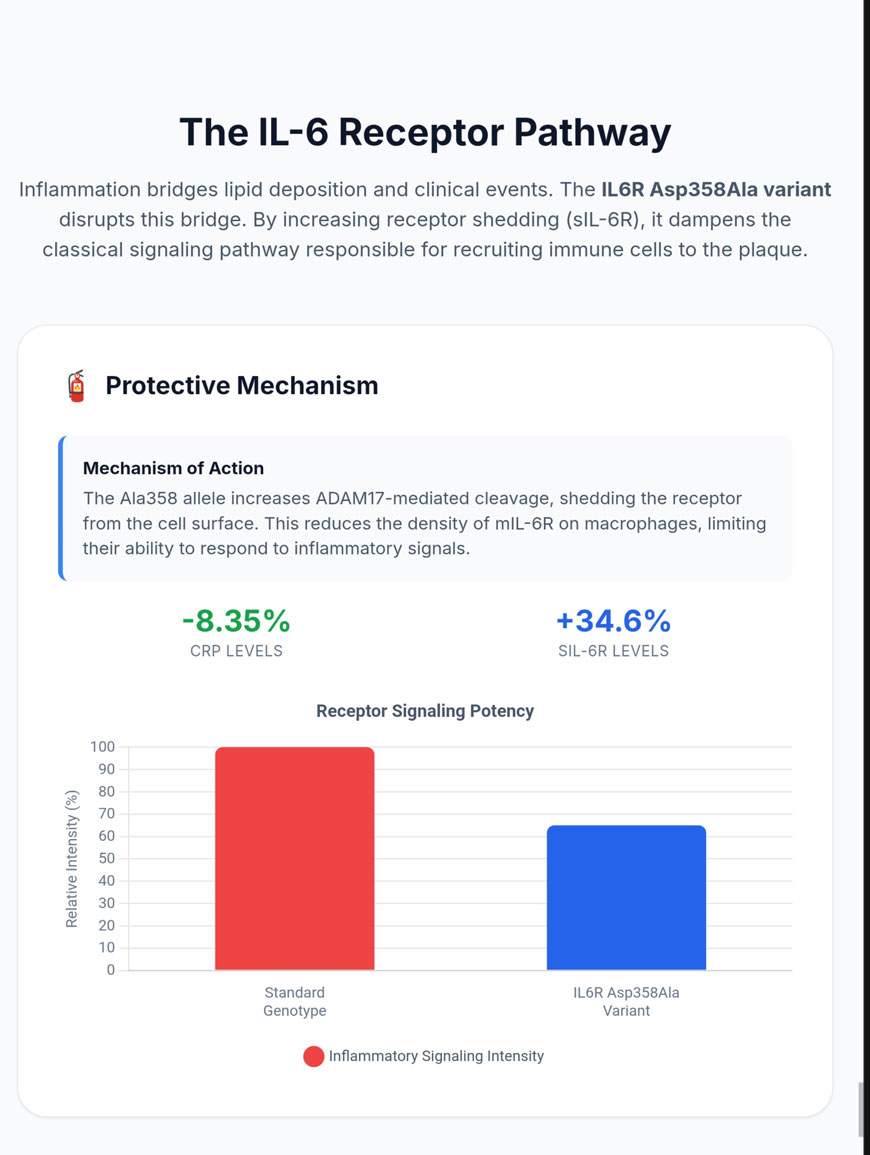
The Asp358Ala Variant and the Shedding Mechanism
The identification of the rs2228145 single nucleotide polymorphism (SNP) in the IL6R gene, resulting in a p.Asp358Ala substitution, has provided a unique genetic lens through which to observe the causal role of IL-6 in coronary heart disease [1]. This non-synonymous variant involves a transition from adenine to cytosine at position 1073 of the gene, located within exon 9 which encodes the extracellular domain of the receptor [2]. The substitution occurs in close proximity to the cleavage site (Valine 356) recognized by the metalloproteinase ADAM17, also known as the “sheddase” responsible for converting mIL-6R into sIL-6R [2].
The presence of the alanine (C) allele significantly enhances the susceptibility of the receptor to proteolytic cleavage, leading to a profound shift in receptor distribution. Carriers of the Ala358 variant exhibit a marked reduction in the surface expression of mIL-6R on monocytes and CD4+ T cells—up to a 28% reduction per allele—while simultaneously showing a substantial increase in circulating sIL-6R levels, typically a 34.6% increase per copy of the minor allele [2].
Mendelian Randomization and Causal Protection Statistics
Using the Asp358Ala variant as a genetic instrument for Mendelian randomization allows for the estimation of the long-term effects of IL-6 receptor inhibition in human populations [1]. The evidence consistently demonstrates that the dampening of IL-6 signaling via this mechanism provides a protective effect against a spectrum of cardiovascular and inflammatory diseases [1–3]. By comparing the genetic findings with the effects observed in randomized trials of the IL-6R monoclonal antibody tocilizumab, researchers have confirmed that the genetic variant effectively mimics pharmacological blockade [1].
Table 1. Phenotype or Biomarker Associations
| Phenotype or Biomarker | Effect Size (per Ala358 Allele) | 95% Confidence Interval | p-values |
| IL-6R Plasma Concentration | +34.6% (relative increase) | [33.2%, 36.1%] | < 1 × 10⁻²⁰ |
| C-Reactive Protein (CRP) | -8.35% (relative decrease) | [-9.38%, -7.31%] | < 1 × 10⁻¹⁵ |
| Fibrinogen Concentration | -0.85% (relative decrease) | [-1.10%, -0.60%] | 1.2 × 10⁻¹⁰ |
| Coronary Heart Disease (CHD) | 0.95 (Odds Ratio) | [0.93, 0.97] | 1.53 × 10⁻⁵ |
| Peripheral Artery Disease (PAD) | 0.91 (Odds Ratio) | [0.88, 0.94] | 6.2 × 10⁻⁹ |
Data synthesized from research consortia analyzing up to 133,449 individuals [1].
The statistical significance of these findings suggests that IL-6R signaling is not merely a marker of disease but a causal participant. The reduction in CAD risk occurs despite the potential increase in the half-life of circulating IL-6 molecules bound to the sIL-6R; the net biological effect is a reduction in the inflammatory response needed to build and destabilize plaque [1]. This genetic “dampening” prevents the artery from mounting the massive inflammatory response typically associated with lipid retention, effectively decoupling the presence of LDL from the development of clinical disease. Recent studies in the Bangladeshi population further support that this polymorphism is associated with a reduced risk of type 2 diabetes and hypertension [5].
Therapeutic Implications and Broader Protective Profiles
The insights derived from the Asp358Ala variant have profound implications for drug development. Since the variant reduces systemic inflammation (evidenced by lower CRP and fibrinogen) without significantly altering traditional risk factors like blood pressure, it highlights IL-6R blockade as a precision tool for mitigating residual inflammatory risk [1]. Furthermore, the protective effects of this variant extend beyond chronic cardiovascular disease to acute critical illnesses. Mendelian randomization studies have indicated that IL-6R blockade is causally associated with reduced incidence of sepsis and improved outcomes in severe COVID-19 [4].
However, the mechanism is not entirely without trade-offs. While the Asp358Ala allele protects against CAD and rheumatoid arthritis, it may increase susceptibility to certain infections, mirroring the side-effect profile of pharmacological IL-6R antagonists [2]. This highlights the necessity of a nuanced understanding of the IL-6/IL-6R axis, where the variant acts as a “natural experiment” in balancing inflammatory protection against host defense.
MIA3 and the Structural Integrity of the Fibrous Cap
While inflammatory modulation prevents the initiation of plaque, the stability of a lesion once formed is governed largely by the structural characteristics of the vessel wall. The MIA3 gene, encoding the protein TANGO1 (Transport and Golgi Organization 1), has emerged as a cornerstone of vascular resilience through its role in the secretion of extracellular matrix components [6].
TANGO1 Biochemistry and Cargo Export
TANGO1 is an evolutionarily conserved protein resident in the endoplasmic reticulum (ER) membrane. Its primary function is to facilitate the export of bulky secretory cargoes that are too large to fit into standard COPII vesicles, which typically have a diameter of only 60–80 nm [6]. Large molecules, such as procollagen—which can reach lengths of 300 nm—require a specialized transport mechanism. TANGO1 achieves this by organizing into a ring-like scaffold at ER exit sites (ERES) and recruiting ER-Golgi intermediate compartment (ERGIC) membranes to create a transient, enlarged secretory tunnel [6].
The protein utilizes a luminal SH3-like domain to bind specifically to triple-helical collagen motifs and the chaperone HSP47, ensuring that only properly folded proteins are packaged for export [6]. This machinery is essential for the transport of several collagen classes, including interstitial fibrillar collagens (I, II, and III), non-fibrillar basement membrane collagen (IV), and FACIT collagens (IX) [6].
The rs67180937 Variant and VSMC Phenotypic Stability
The genetic regulation of MIA3 expression in the vasculature is critical for determining whether a plaque becomes a dangerous, rupture-prone lesion or a stable “scar.” Genome-wide association studies have identified a locus on chromosome 1q41 where the G allele of the lead SNP rs67180937 is associated with an increased risk of coronary artery disease [7]. Mechanistic studies have revealed that this risk allele is correlated with lower expression of MIA3 in vascular smooth muscle cells (VSMCs) and a significantly reduced proliferative response to growth factors [7].
In a resilient vascular phenotype, high MIA3 expression promotes the transition of VSMCs toward a protective, synthetic phenotype. These cells migrate to the intima and produce the collagen-rich extracellular matrix required to build a thick, fibrous cap over the lipid core of a plaque [7].
Table 2. Plaque Characteristic by MIA3 Expression
| Plaque Characteristic | Low MIA3 Expression (Risk Genotype) | High MIA3 Expression (Protective Genotype) |
| VSMC Proliferation | Reduced; fewer cells in the cap | Enhanced; robust cellular presence |
| Collagen Deposition | Compromised; thin, fragile matrix | Efficient; thick, protective fibrous cap |
| Clinical Outcome | Rupture-prone “vulnerable” plaque | Stable, asymptomatic “scarred” plaque |
Data synthesized from studies on human donor VSMCs and murine models [7].
The relevance of MIA3 is further underscored by observations in human coronary artery lesions, where a significant reduction in MIA3 protein abundance is seen in the thin-cap regions of unstable plaques compared to the thick caps of stable lesions [7].
SCARB1 and the Gatekeeping of Lipoprotein Transcytosis
A critical early step in atherogenesis is the physical entry of low-density lipoprotein (LDL) into the subendothelial space. It is now recognized as a highly regulated transcellular transport process known as transcytosis, primarily mediated by the scavenger receptor class B type 1 (SR-B1), encoded by the SCARB1 gene [8].
The DOCK4/Rac1 Signaling Axis
The mechanism by which endothelial SR-B1 internalizes and transports LDL particles involves a complex signaling cascade. Upon the binding of LDL to the extracellular domain of SR-B1, a specific cytoplasmic domain of the receptor recruits the guanine nucleotide exchange factor DOCK4 (Dedicator of Cytokinesis 4) [8]. This recruitment is a prerequisite for the activation of the small GTPase Rac1, which orchestrates the actin cytoskeleton rearrangements necessary for transport [8].
Table 3. Molecular Component Function in LDL Transcytosis
| Molecular Component | Function in LDL Transcytosis |
| SR-B1 (SCARB1) | Primary receptor for LDL capture at the luminal surface |
| IQAYSESL Motif | Cytoplasmic docking site for adapter proteins |
| DOCK4 | Guanine nucleotide exchange factor; recruits to SR-B1 |
| Rac1 | GTPase that triggers actin-mediated internalization |
Data synthesized from endothelial cell transport studies [8].
Experimental evidence using mice with endothelial-specific deletion of SR-B1 has shown a 60–80% reduction in LDL delivery into the artery wall, leading to a massive decrease in atherosclerotic lesion area [8]. This indicates that variants in SCARB1 that lower the rate of this transcytosis can fundamentally arrest the development of atherosclerosis at the “entry” stage [8].
Human SCARB1 Variants and the Lipoprotein Paradox
In the liver, SR-B1 is the principal receptor for the selective uptake of cholesteryl esters from HDL. Consequently, rare loss-of-function variants in SCARB1 can lead to extremely high levels of circulating HDL cholesterol, yet these individuals often face increased coronary risk because reverse cholesterol transport is impaired [9,10].
Table 4. SCARB1 Genetic Variant Impact
| SCARB1 Genetic Variant | Impact on Lipid Profile | Impact on Vascular Risk |
| rs5888 (TT Genotype) | Associated with higher HDL-C | Often protective (especially in non-Asian males) |
| rs144334493 (Deletion) | Attenuates FOXA1 binding; lowers SR-B1 | Increased susceptibility to CHD |
| S129L Substitution | Combined High HDL-C and High Lp(a) | Diminished Lp(a) clearance; high risk |
Data synthesized from meta-analyses and cohort studies [10].
Resilience is associated with the efficiency of the receptor’s function in both the liver and the endothelium. Favorable polymorphisms that lower endothelial transcytosis without compromising hepatic reverse cholesterol transport represent the ideal “super-resilient” genotype [8–10].
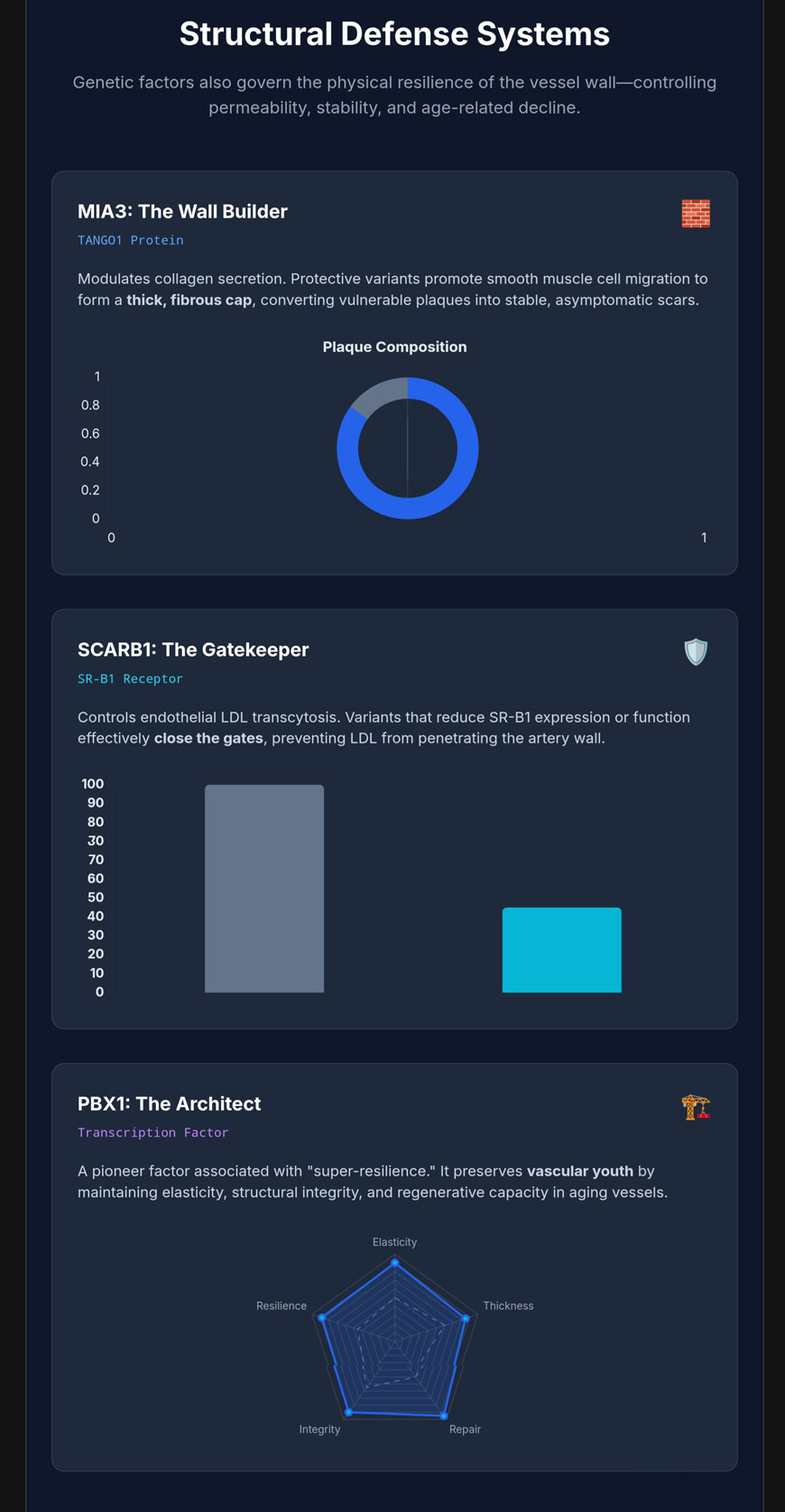

PBX1 and the Preservation of Vascular Youth
Vascular aging is a precursor to many forms of cardiovascular disease. Recent studies have identified variants near the PBX1 (Pre-B-cell leukemia homeobox 1) gene as being associated with exceptional vascular health in older adults [11].
PBX1 as a Pioneer Transcription Factor
PBX1 belongs to the TALE (Three Aminoacid Loop Extension) class of homeobox transcription factors and functions as a “pioneer factor,” meaning it can access closed chromatin and mark specific genes for transcriptional activation [12,13]. In the cardiovascular system, PBX1 coordinates transcriptional pathways that control aortic patterning and the development of the cardiac outflow tract [12,13]. In the aging adult vasculature, PBX1 expression is essential for the repair of endothelial damage and the maintenance of endothelial progenitor cell populations [11–13].
Epigenetic Regulation and Vascular Resilience
The resilience associated with PBX1 variants likely stems from their ability to preserve the youthful epigenetic state of the vessel wall. By maintaining high expression of repair genes and regulating cellular oxidative stress and apoptosis, PBX1 prevents structural decay [12,13].
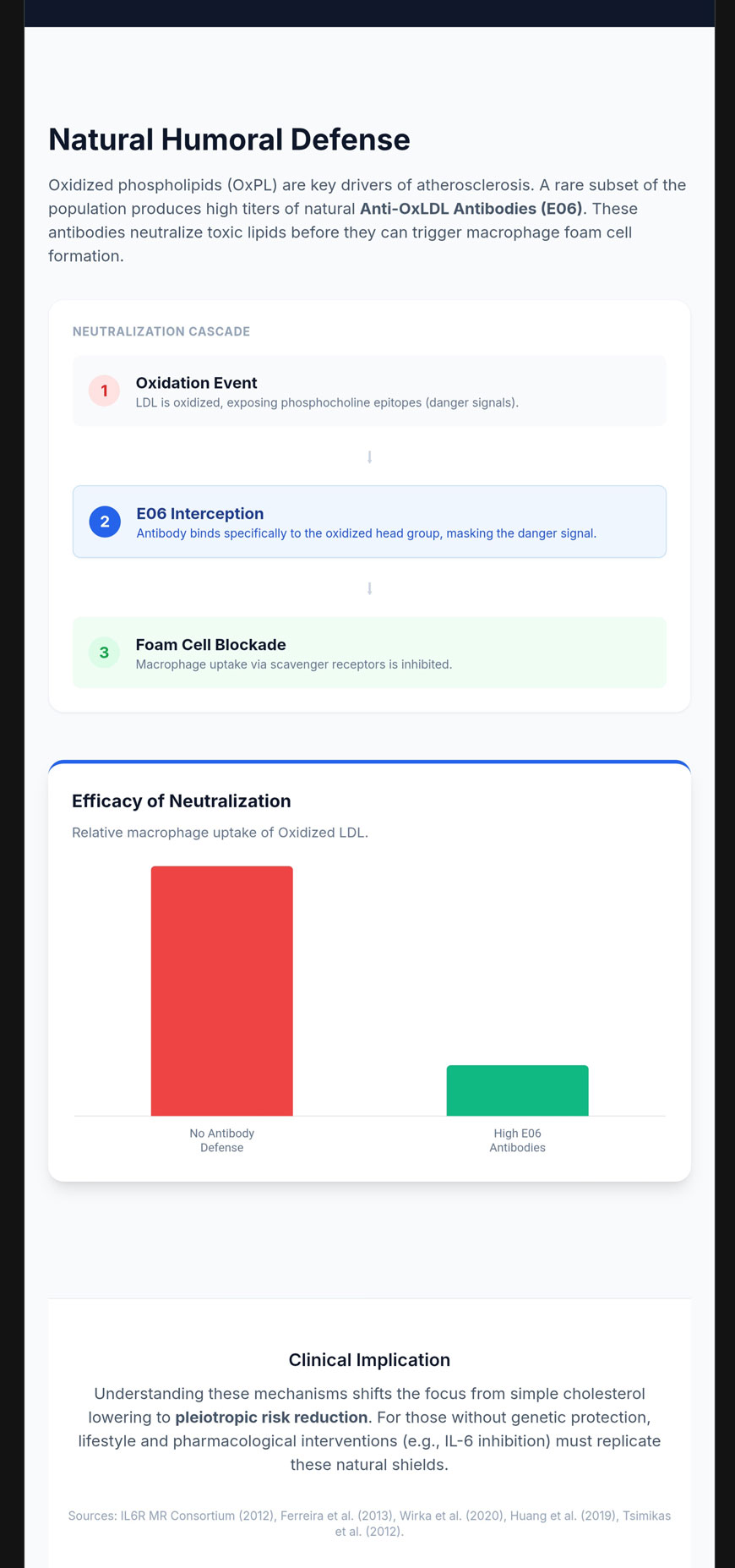
Natural Anti-OxLDL Antibodies and Humoral Defense
Even when lipoproteins enter the vessel wall and become oxidized (OxLDL), they can be neutralized by naturally occurring autoantibodies like E06 before they trigger the inflammatory foam cell cycle [15].
The E06 Antibody and the Phosphocholine Epitope
E06 is a natural IgM autoantibody that specifically binds to the phosphocholine (PC) headgroup of oxidized phospholipids (OxPL) [15]. Upon oxidation of the sn-2 fatty acid fragment, the PC headgroup becomes exposed as a neoepitope or “danger signal” [15].
Table 5. Biological Entity Recognition by E06
| Biological Entity | Recognition by E06 | Mechanism of Action |
| Native LDL | No | Avoids interference with normal lipid metabolism |
| Copper-Oxidized LDL | Yes | Binds to fragmented OxPL on the surface |
| Lipoprotein(a) | Yes | Neutralizes OxPL covalently bound to Apo(a) |
| Apoptotic Cells | Yes | Recognizes exposed PC on damaged membranes |
Data synthesized from immunological assays and structural studies [15].
The protective mechanism of E06 is twofold: it inhibits the uptake of these particles by macrophage scavenger receptors, preventing foam cell formation, and neutralizes pro-inflammatory signaling via receptors like TLR-2 and TLR-4 [15].
Therapeutic Potential and Clinical Correlates
The existence of high natural levels of E06-like antibodies provides a robust “innate immunization.” This has prompted the development of therapeutic candidates like VB-201, a small-molecule lecinoxoid designed to compete with pro-inflammatory OxPLs for binding to CD14 and TLR-2 [16]. High levels of OxPL-apoB (measured via E06-based ELISA) are powerful biomarkers for predicting the presence and progression of coronary, femoral, and carotid artery disease [14].
References
- Interleukin-6 Receptor Mendelian Randomisation Analysis (IL6R MR) Consortium, Swerdlow DI, Holmes MV, et al. The interleukin-6 receptor as a target for prevention of coronary heart disease: a mendelian randomisation analysis. Lancet. 2012;379(9822):1214-1224. doi:10.1016/S0140-6736(12)60110-X
- Ferreira RC, Freitag DF, Cutler AJ, et al. Functional IL6R 358Ala allele impairs classical IL-6 receptor signaling and influences risk of diverse inflammatory diseases. PLoS Genet. 2013;9(4):e1003444. doi:10.1371/journal.pgen.1003444
- Levin MG, Klarin D, Georgakis MK, et al. A Missense Variant in the IL-6 Receptor and Protection From Peripheral Artery Disease. Circ Res. 2021;129(10):968-970. doi:10.1161/CIRCRESAHA.121.319589
- Hamilton FW, Thomas M, Arnold D, et al. Therapeutic potential of IL6R blockade for the treatment of sepsis and sepsis-related death: A Mendelian randomisation study. PLoS Med. 2023;20(1):e1004174. Published 2023 Jan 30. doi:10.1371/journal.pmed.1004174
- Aparicio-Siegmund S, Garbers Y, Flynn CM, et al. The IL-6-neutralizing sIL-6R-sgp130 buffer system is disturbed in patients with type 2 diabetes. Am J Physiol Endocrinol Metab. 2019;317(2):E411-E420. doi:10.1152/ajpendo.00166.2019
- Wilson DG, Phamluong K, Li L, et al. Global defects in collagen secretion in a Mia3/TANGO1 knockout mouse. J Cell Biol. 2011;193(5):935-951. doi:10.1083/jcb.201007162
- Aherrahrou R, Guo L, Nagraj VP, et al. Genetic Regulation of Atherosclerosis-Relevant Phenotypes in Human Vascular Smooth Muscle Cells. Circ Res. 2020;127(12):1552-1565. doi:10.1161/CIRCRESAHA.120.317415
- Huang L, Chambliss KL, Gao X, et al. SR-B1 drives endothelial cell LDL transcytosis via DOCK4 to promote atherosclerosis. Nature. 2019;569(7757):565-569. doi:10.1038/s41586-019-1140-4
- Hu S, Hu D, Wei H, et al. Functional Deletion/Insertion Promoter Variants in SCARB1 Associated With Increased Susceptibility to Lipid Profile Abnormalities and Coronary Heart Disease. Front Cardiovasc Med. 2022;8:800873. Published 2022 Jan 13. doi:10.3389/fcvm.2021.800873
- Ye LF, Zheng YR, Zhang QG, Yu JW, Wang LH. Meta-analysis of the association between SCARB1 polymorphism and fasting blood lipid levels. Oncotarget. 2017;8(46):81145-81153. Published 2017 Sep 14. doi:10.18632/oncotarget.20867
- Wen Y, Chen H, Wang Y, et al. Extracellular vesicle-derived TP53BP1, CD34, and PBX1 from human peripheral blood serve as potential biomarkers for the assessment and prediction of vascular aging. Hereditas. 2024;161(1):3. Published 2024 Jan 3. doi:10.1186/s41065-023-00306-8
- Crisafulli L, Brindisi M, Liturri MG, Sobacchi C, Ficara F. PBX1: a TALE of two seasons-key roles during development and in cancer. Front Cell Dev Biol. 2024;12:1372873. Published 2024 Feb 9. doi:10.3389/fcell.2024.1372873
- Chen H, Yu Z, Niu Y, Wang L, Xu K, Liu J. Research progress of PBX1 in developmental and regenerative medicine. Int J Med Sci. 2023;20(2):225-231. Published 2023 Jan 22. doi:10.7150/ijms.80262
- Taleb A, Witztum JL, Tsimikas S. Oxidized phospholipids on apoB-100-containing lipoproteins: a biomarker predicting cardiovascular disease and cardiovascular events. Biomark Med. 2011;5(5):673-694. doi:10.2217/bmm.11.60
- Hörkkö S, Bird DA, Miller E, et al. Monoclonal autoantibodies specific for oxidized phospholipids or oxidized phospholipid-protein adducts inhibit macrophage uptake of oxidized low-density lipoproteins. J Clin Invest. 1999;103(1):117-128. doi:10.1172/JCI4533
- Mendel I, Feige E, Yacov N, et al. VB-201, an oxidized phospholipid small molecule, inhibits CD14- and Toll-like receptor-2-dependent innate cell activation and constrains atherosclerosis. Clin Exp Immunol. 2014;175(1):126-137. doi:10.1111/cei.12212