
The Great Cholesterol Debate: Why Your “Healthy” Diet Might Be Sending Your LDL Through the Roof
Imagine you have just started a new way of eating. You have cut out bread, pasta, and sugar. Instead, you are eating more healthy fats, like steak, eggs, and butter. You feel better than you have in years. Your “sugar levels” are great, your blood pressure is low, and you have plenty of energy. In your mind, you are “metabolically perfect.” This means your body is doing a great job of handling energy and keeping you fit.
You go to your doctor for a check-up, expecting them to be impressed. But when the blood test results come back, your doctor looks worried. Your LDL level—the one most people call “bad cholesterol”—is through the roof. It is not just a little high; it looks like a “red alert” on the computer screen.
This is a very common “medical mystery” today. Many people who follow a “keto” or low-carb diet find themselves in this spot. They feel like a million bucks, but their blood work says they are at risk for a heart attack. How can someone who is so healthy on the outside have such scary numbers on the inside?
Right now, the world of science is split into two camps. There is the “old school” group of heart doctors. They believe that high LDL is always like a toxin in your blood. Then there is a “new school” group of thinkers. They believe that if you are lean and fit, high LDL might just be a sign that your body is moving energy around in a different way.
To help you understand this deep debate, we are going to look at the seven biggest takeaways from the latest science. We will use “cautious metabolic respect.” This means we will respect your healthy lifestyle while also being careful about what the numbers might be telling us.
Takeaway 1: The “Cookie vs. Statin” Surprise
One of the most interesting stories in this debate involves a box of Oreo cookies. It sounds like a joke, but it was a real experiment by a researcher named Nick Norwitz. He wanted to show that the way our bodies react to food is not the same for everyone.
He took a group of lean, healthy people who were on a keto diet and had very high LDL. He gave them a choice: take a statin (a drug that lowers cholesterol) or eat a pack of Oreo cookies every day for a short time.
The result was shocking. Eating the cookies actually lowered their LDL more than the medicine did!
Why did this happen? It all comes down to your liver and its “sugar tanks.” When you don’t eat carbs, your sugar tanks (called glycogen) stay empty. Your liver sees this and decides to send out more fat to feed your muscles. When you eat the cookies, you fill those sugar tanks. The liver sees the sugar and tells the “fat delivery buses” to stay home.
This experiment is what scientists call “legit bait.” It is used to show that the current medical rules might not fit everyone. As the source says:
“By demonstrating that a processed cookie can lower LDL-C more effectively than a statin in a specific metabolic context, they raise the burden of proof for the ‘LDL-as-toxin’ narrative.”
Takeaway 2: LDL Isn’t Just “Gunk”—It’s a Delivery Bus
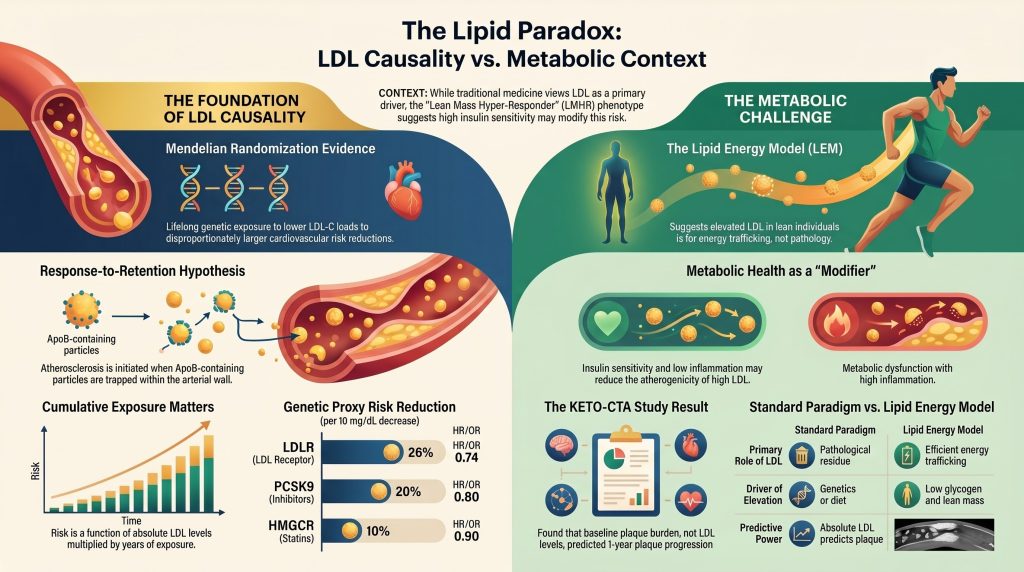
To understand the “new school” view, you need to know about the Lipid Energy Model (LEM). Most people think LDL is just “trash” or “gunk” left over in the blood that clogs your heart. But the LEM says LDL is actually more like a “delivery bus.”
When your “sugar tanks” are low because you aren’t eating carbs, your body needs fuel. Your liver packages up fats into little buses called VLDL. As these buses drop off their fat fuel to your muscles, they turn into LDL.
In people who are very lean and fit—often called “Lean Mass Hyper-Responders”—the body is just very good at moving this fat energy around. In this view, high LDL isn’t a sign of disease. It’s a sign that the delivery buses are working overtime to keep your muscles moving.
| Component | Standard View (Old School) | Energy Model (New School) |
| What is LDL? | It is trash or residue that causes disease. | It is a bus that delivers fat fuel to the body. |
| Does context matter? | Risk is high no matter how fit you are. | High LDL is safe if you are healthy and lean. |
| Why is it high? | Bad genes or eating too much animal fat. | Low sugar tanks and a very lean body. |
| The Role of ApoB | ApoB is a “magnetic hook” that makes the bus stick to your heart walls. | ApoB is just a part of the bus and is not a problem if you are fit. |
Takeaway 3: The Elderly Paradox (Why High LDL Might Help at 80)
There is a strange finding in science called the “Elderly Paradox.” Studies often show that people over 60 or 70 who have high LDL live just as long—or even longer—than those with low LDL.
This happens because LDL has a secret job: it helps your immune system. LDL particles act like little sponges that grab onto germs and stop them from making you sick. In older people, fighting off an infection like the flu or pneumonia is a huge part of staying alive. In these cases, the “protective” power of LDL might be more important than the risk of heart disease.
However, we have to be careful. There are two “biases” that can trick us here:
- Reverse Causation: People who are very sick, like with cancer, often see their cholesterol drop right before they pass away. This makes low cholesterol look “bad” when it was actually just a sign of being very sick.
- Survivor Bias: The people who make it to age 80 with high LDL might just be “hardened survivors.” Maybe the people who were sensitive to LDL already had heart trouble years ago, leaving behind only the people whose bodies can handle it.
Takeaway 4: The “Bricks in the Wall” Problem
Even if you are the healthiest person in the gym, many doctors are still worried about high LDL. They use a rule called the “response-to-retention” model. Think of your artery wall like a brick wall, and think of LDL particles as the “bricks.”
In this model, you cannot build a “clog” (plaque) without bricks. Even if you are a “world-class builder” with perfect metabolic health, if you have five times more bricks than you need, it is much more likely that some will get stuck where they don’t belong.
This brings us to a special part of the LDL bus called ApoB. You can think of ApoB as a “magnetic hook” on the outside of the bus. This hook is “positively charged,” while your artery wall is “negatively charged.” They stick together like magnets.
“The biological mechanism for LDL causality is established through the ‘response-to-retention’ model, which identifies the subendothelial entrapment of ApoB-containing lipoproteins as the necessary initiating event of atherosclerosis.”
Here is the simple step-by-step of how the “clog” starts:
- The Trap: The buses (LDL) get stuck to the wall because of the “magnetic hook” (ApoB).
- The Mess: Once stuck, the particles change and “rot” (this is called oxidation).
- The Clean-up: Your body sends “cleaner cells” to eat the mess. But the cleaners eat too much, get stuck, and die.
- The Wall: Over 20 or 30 years, these dead cleaners and trapped bricks turn into a hard wall of plaque.
Takeaway 5: Your Genes Are a Time Machine
How do we know for sure that LDL is the “brick” that causes the problem? Scientists use something called Mendelian Randomization. This is like a “lifelong trial” you are born into.
Some people are born with lucky genes that keep their LDL very low for their entire lives. When scientists look at these people, they find they have a huge reduction in heart risk. Here are the numbers from the source:
- People with a lucky LDLR gene have a 26% lower risk of heart trouble.
- People with a lucky PCSK9 gene have a 20% lower risk.
- Even taking a drug like a statin later in life only gives a 10% lower risk.
This tells us that “cumulative exposure”—or how much LDL you have over many years—is what matters most. Think of it like smoking. One cigarette won’t give you lung cancer. It is the “area under the curve”—the total amount of smoke your lungs see over 40 years—that causes the disease.
“The magnitude of risk reduction per unit of LDL-C lowering in MR studies is approximately three times greater than that seen in statin trials. This highlights the ‘cumulative exposure’ principle: the risk of ASCVD is a function of both the absolute level of ApoB particles and the duration of exposure.”
Even if you feel great now, having very high LDL for 20 years might be like smoking a pack a day for 20 years. The damage adds up slowly over time.
Takeaway 6: The “Plaque Begets Plaque” Warning (KETO-CTA)
A new study called KETO-CTA looked at 100 people on keto diets who had very high LDL. They scanned their hearts to see what was happening.
At first, the “new school” group was excited. They found that 57% of the people had a “Zero CAC” score, which means they had no hard, old plaque in their hearts. This made it look like they were safe.
However, when the scientists looked closer, they saw a major red flag. They found that “soft plaque” (the kind that is more likely to cause a sudden heart attack) grew by about 20% in just one year. This is 3 to 5 times higher than what we see in high-risk groups like diabetics.
This study taught us that “plaque begets plaque.” This means if you already have a little bit of a clog, the high LDL will make it grow much faster. Even if you are fit and healthy, your body might not be able to stop the flood if the “water” (the LDL bricks) stays too high for too long.
Takeaway 7: The “Truth Sandwich” on LDL and Immunity
You will often hear people online say that high LDL is actually “good” because it helps your immune system. Let’s look at the “Truth Sandwich” to find the real answer:
- Fact: It is true that LDL particles help your body fight off germs and infections. They are a key part of your body’s defense system.
- The Nuance: Just because LDL helps your immune system does not mean it is safe for your heart. Both things can be true at the same time.
- Fact: High levels of LDL for too many years still lead to particles getting trapped in your artery walls. Even a strong, clean body cannot always stop a flood if the water level stays too high for too long. This trapping is what starts heart disease.
Conclusion: Cautious Respect for the Unknown
The debate over cholesterol is not a simple fight between “good” and “bad.” It is about having “cautious metabolic respect.”
We must respect that eating a healthy diet and being fit makes you much safer than someone who is unhealthy. Having low inflammation and good blood pressure is like having “better mortar” for your wall. However, we must also respect the mountain of evidence showing that LDL is the “brick” that builds the clog. Being fit might help you, but it might not be a “suit of armor” that protects you forever.
“Individualized risk assessment… recognizes that ‘excellent metabolic health’ may not be a suit of armor against the long-term effects of extreme hyperlipidemia.”
As you look at your own health, ask yourself this: Are you comfortable basing your long-term life on a new theory that hasn’t been proven over 20 years? Or is it time to look closer at the “bricks” in your own wall?
The best path is to work with a doctor who understands both sides. You want someone who celebrates your great metabolic health but also keeps a close eye on those “delivery buses” to make sure they aren’t getting stuck where they don’t belong.
DEEP DIVE
The Causal Role of Low-Density Lipoprotein in Atherosclerotic Cardiovascular Disease: Evidence Synthesis, Metabolic Context, and Scientific Epistemology
Abstract
The identification of low-density lipoprotein (LDL) as a causal agent in atherosclerotic cardiovascular disease (ASCVD) represents one of the most rigorously tested paradigms in modern medicine. This conclusion is supported by converging evidence from lifelong genetic exposure studies using Mendelian randomization, large-scale prospective epidemiology, and randomized controlled trials (RCTs). In recent years, an intellectual movement has emerged, primarily within the ketogenic and low-carbohydrate communities, challenging the universality of this causal relationship. This movement is exemplified by the ‘Lean Mass Hyper-Responder’ (LMHR) phenotype and the associated ‘Lipid Energy Model’ (LEM), which propose that in individuals with high insulin sensitivity and lean body mass, elevated LDL-C may not carry equivalent pathological implications. This paper analyzes the epistemological landscape of this debate, evaluates the strength of evidence on both sides, and proposes a framework for productive clinical communication with metabolically informed skeptics.
1. Introduction
The existence of skepticism regarding LDL’s causal role in ASCVD frequently arises from legitimate inquiry into the limitations of population-level data when applied to unique metabolic contexts. Healthy scientific skepticism serves as a necessary mechanism for paradigm refinement, ensuring that models account for edge cases and anomalous data. Within lipidology, this skepticism is often catalyzed by the observation of high-functioning, metabolically healthy individuals who exhibit lipid profiles that traditional guidelines would classify as high-risk.
Distinguishing between healthy skepticism, contrarianism, and conspiracy-style reasoning is essential for productive dialogue. Healthy skepticism seeks to expand models to include new data. Contrarianism defines itself in opposition to institutional consensus, often prioritizing a single contradictory data set over the totality of the field. Conspiracy-style reasoning goes further, suggesting that the consensus is intentionally deceptive, sometimes citing pharmaceutical industry influence on clinical guidelines. Labeling all dissent as ‘misinformation’ can be counterproductive, as it alienates intelligent observers who perceive such labeling as an appeal to authority rather than transparent engagement with underlying data.
2. The Persuasiveness of Contemporary Skeptical Arguments
Arguments presented by proponents of the Lipid Energy Model resonate with a specific audience because they provide a sophisticated, mechanistic narrative that addresses the felt experience of individuals on carbohydrate-restricted diets (CRDs). Their communication strategies are characterized by several distinct patterns that build internal consistency within their models.
2.1 The Lipid Energy Model and Metabolic Context
The cornerstone of modern LDL skepticism is the transition from evaluating LDL-C as an isolated risk factor to assessing it within a broader metabolic framework. Skeptics emphasize markers of insulin sensitivity—such as a low triglyceride to HDL-C ratio—arguing that high-quality metabolic markers render elevated LDL-C benign. The Lipid Energy Model (LEM) provides the mechanistic rationale, proposing that in lean individuals with low hepatic glycogen, the liver upregulates export of Very Low-Density Lipoprotein (VLDL) to traffic triglycerides to peripheral tissues, leading to elevated LDL-C as a downstream consequence of efficient lipid metabolism rather than dyslipidemia in the classical sense.
Table 1. Comparison of the Standard Lipid Paradigm and the Lipid Energy Model
| Component | Standard Lipid Paradigm | Lipid Energy Model (LEM) | Key Distinction |
| Primary Role of LDL | Pathological residue of lipid transport. | Result of efficient energy trafficking via VLDL turnover. | Mechanism vs. pathology distinction. |
| Contextual Modifier | Risk is additive with other cardiovascular risk factors. | Risk is context-dependent; proposed to be benign if insulin sensitive. | Population vs. individual risk framing. |
| Primary Driver of Elevation | Genetic defect (e.g., FH) or high saturated fat intake. | Low hepatic glycogen and lean mass driving upregulated VLDL export. | Metabolic state as a proposed modulator. |
| Predictive Power | Absolute ApoB/LDL-C predicts atherosclerotic plaque burden. | Baseline plaque burden predicts progression; circulating lipid levels may be secondary. | Disputed by longitudinal data lacking an adequate control group. |
This model is particularly persuasive because it offers testable predictions, such as an inverse association between BMI and LDL-C increase on a ketogenic diet. When intelligent skeptics observe these predictions confirmed in small cohorts or self-experiments, it reinforces the belief that the mainstream model is incomplete or misapplied to their phenotype.
2.2 Rhetorical Framing and the Burden of Proof
Skeptical communicators often highlight perceived absurdities in current guidelines through dramatic experiments designed to reveal discordance between LDL-C and other metabolic markers. By demonstrating that a specific dietary or pharmacological intervention can lower LDL-C in a metabolically neutral context, they raise the evidentiary burden for the ‘LDL-as-toxin’ narrative. Furthermore, they highlight discordance between risk markers—such as a zero Coronary Artery Calcium (CAC) score in the presence of very high LDL-C—to question the predictive validity of population-level risk equations in individual cases.
3. The Robust Case for LDL Causality: Convergence of Evidence
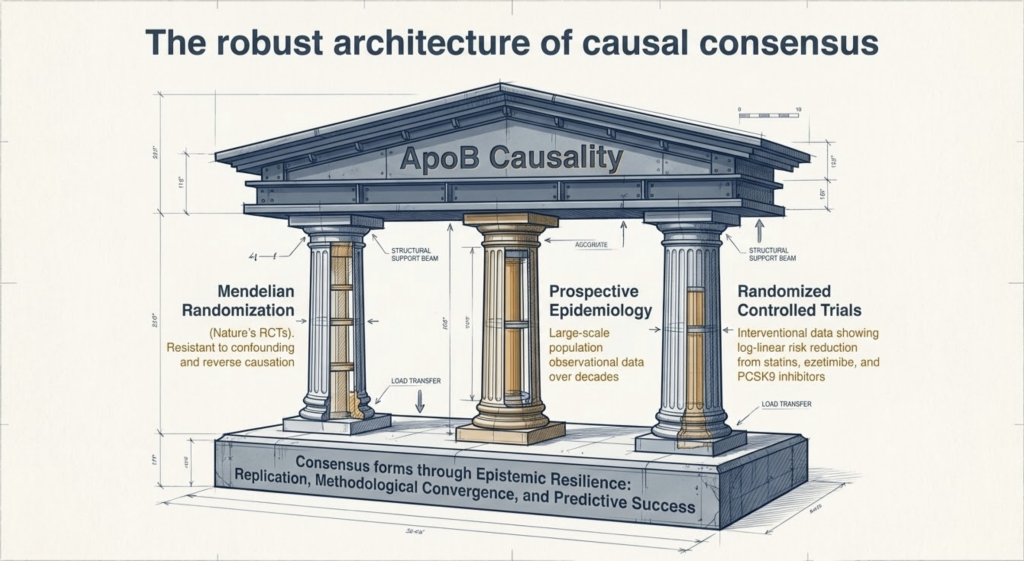
The scientific consensus that LDL is causal in ASCVD is not built on a single study but on the convergence of independent lines of evidence that collectively satisfy the Bradford Hill criteria for causality. This convergence is critical because each method possesses different strengths and limitations, yet they all point to the same conclusion: cumulative exposure to ApoB-containing lipoproteins is the primary determinant of atherosclerotic development.
3.1 Genetic Evidence and Mendelian Randomization
Mendelian randomization (MR) provides perhaps the strongest evidence for causality by utilizing the random assortment of genetic variants at conception to mimic a lifelong randomized controlled trial, largely resistant to confounding and reverse causation. Variants in genes encoding the LDL receptor (LDLR), PCSK9, HMG-CoA reductase (HMGCR), NPC1L1, and APOB all affect LDL-C through distinct biological pathways, yet they consistently demonstrate that lower lifelong exposure to LDL-C leads to a disproportionately larger reduction in cardiovascular risk compared to short-term pharmacological interventions initiated later in life.
Table 2. Mendelian Randomization Estimates of Cardiovascular Risk Reduction by Genetic Proxy (per 10 mg/dL decrease in LDL-C)
| Genetic Proxy (Drug Target) | Risk Reduction per 10 mg/dL LDL-C Decrease | Hazard/Odds Ratio (95% CI) |
| LDLR (LDL Receptor / FH model) | 26% | 0.74 (0.66–0.82) |
| PCSK9 (PCSK9 Inhibitor proxy) | 20% | 0.80 (0.75–0.86) |
| HMGCR (Statin proxy) | 10% | 0.90 (0.86–0.94) |
| NPC1L1 (Ezetimibe proxy) | 15% | 0.85 (0.79–0.91) |
The magnitude of risk reduction per unit of LDL-C lowering in MR studies is approximately three times greater than that observed in statin trials of similar LDL-C reduction. This highlights the ‘cumulative exposure’ principle: the risk of ASCVD is a function of both the absolute level of ApoB particles and the duration of exposure. This ‘area under the curve’ concept explains why individuals with familial hypercholesterolemia (FH) develop premature disease, while those with genetically determined low LDL-C are protected throughout their lives.
3.2 Pathophysiology: The Response-to-Retention Model
The biological mechanism for LDL causality is established through the ‘response-to-retention’ model, which identifies the subendothelial entrapment of ApoB-containing lipoproteins as the necessary initiating event of atherosclerosis. The process begins with the movement of LDL and other ApoB-containing particles across the arterial endothelium via transcytosis. Once in the arterial intima, positively charged residues on the apolipoprotein B-100 protein interact with negatively charged glycosaminoglycan (GAG) chains of extracellular matrix proteoglycans, particularly biglycan and versican.
Table 3. Phases of Atherogenesis According to the Response-to-Retention Model
| Phase | Pathological Event | Mechanism | Key Evidence |
| Initiation | Lipoprotein Retention | ApoB-100 binding to intimal proteoglycans (biglycan, versican) via electrostatic interaction. | Site-directed mutagenesis reducing ApoB-proteoglycan affinity markedly attenuates atherosclerosis in animal models. |
| Modification | Aggregation and Oxidation | Secretory sphingomyelinase, lipolytic enzymes, and ROS modify retained particles. | Modified LDL is markedly more atherogenic than native LDL ex vivo. |
| Inflammatory Response | Monocyte Recruitment and Differentiation | Modified LDL signals endothelial VCAM-1/ICAM-1 upregulation; monocyte-to-macrophage differentiation. | Consistent with the ‘response-to-injury’ hypothesis at the molecular level. |
| Plaque Formation | Foam Cell Accumulation | Macrophages ingest modified LDL via scavenger receptors, becoming foam cells that cannot exit the intima. | Core finding of response-to-retention model (Williams & Tabas, 1995). |
| Progression | Plaque Growth, Calcification, and Rupture Risk | Sustained retention and chronic maladaptive inflammation drive necrotic core formation and cap thinning. | Explains why cumulative LDL exposure (‘cholesterol-years’) predicts events. |
Research utilizing site-directed mutagenesis to create LDL particles with reduced proteoglycan-binding affinity has demonstrated that even under conditions of severe hyperlipidemia, these modified particles cause significantly less atherosclerosis in animal models. This confirms that it is not merely the presence of cholesterol, but the retention of ApoB-containing particles, that drives the atherogenic process—a distinction of fundamental mechanistic importance.
3.3 Randomized Controlled Trials and Clinical Outcomes
The efficacy of LDL-C lowering has been validated across numerous large-scale RCTs involving statins, ezetimibe, and PCSK9 inhibitors. A meta-analysis of 26 randomized trials involving 170,000 participants demonstrated that each 1 mmol/L (approximately 39 mg/dL) reduction in LDL-C produces a consistent ~22% relative reduction in major cardiovascular events. Critically, this relationship is log-linear and holds true even at very low levels of LDL-C, supporting the ‘lower is better’ principle, with no identified threshold below which further reduction loses benefit.
Critiques of these trials often focus on changes in post-2005 trial conduct regulations, suggesting that earlier efficacy estimates were inflated. However, large-scale meta-analyses and trials of newer agents—including PCSK9 inhibitors—have continued to affirm the log-linear relationship between LDL-C reduction and cardiovascular risk reduction across all major contemporary trials.
4. Analysis of Skeptical Counter-Evidence
4.1 The ‘Elderly Paradox’ and Reverse Causation
A common skeptical argument holds that high LDL-C is associated with longevity in the elderly. Systematic reviews of cohort studies in populations aged 60 years and older have found that those with higher LDL-C often survive as long as or longer than those with lower levels. However, this observation is substantially complicated by several well-characterized methodological factors.
First, reverse causation: chronic diseases such as malignancy and terminal infections reliably lower cholesterol levels in the years preceding death, inflating apparent mortality in the low-LDL group. Second, immune function: LDL participates in innate immunity by inactivating microbial pathogens and their toxins; in the elderly, where infection is a leading cause of mortality, this protective role may attenuate all-cause mortality benefits even as atherosclerotic risk accumulates. Third, survivor bias: individuals genetically susceptible to LDL-driven atherosclerosis are more likely to have experienced fatal cardiovascular events earlier in life, leaving an enriched cohort of survivors in older age brackets who are less biologically vulnerable to LDL-mediated atherogenesis.
4.2 The Role of Metabolic Health as a Risk Modifier
Skeptics correctly identify that the absolute cardiovascular risk associated with a given LDL-C level is significantly modified by other factors. High insulin sensitivity, low systemic inflammation (as measured by high-sensitivity C-reactive protein), and optimal blood pressure can lower the probability that a retained LDL particle triggers a maladaptive inflammatory cascade. This does not, however, eliminate the role of LDL as the primary initiating agent of atherosclerosis. Rather, it suggests that some individuals have greater arterial resilience and fewer co-amplifying stressors—modifiers of rate and impact, not of causal mechanism.
5. The KETO-CTA Study: New Data and Its Limitations
The recent publication of longitudinal data from the KETO-CTA trial has become a focal point of the LDL debate. This study followed approximately 100 individuals meeting criteria for the LMHR or ‘near-LMHR’ phenotype over one year, using coronary computed tomography angiography (CCTA) to quantify plaque progression.
5.1 Key Findings
The study reported that baseline plaque burden was the strongest predictor of future plaque progression, whereas traditional lipid markers—including ApoB and LDL-C—and cumulative exposure during a ketogenic diet did not significantly correlate with progression in this cohort over the one-year follow-up. The authors interpreted these findings as evidence that elevated ApoB and LDL-C do not drive atherosclerosis in a dose-dependent manner in metabolically healthy individuals.
Table 4. Key Metrics from the KETO-CTA Cohort and Mainstream Cardiology Context
| Metric | KETO-CTA Cohort Result | Mainstream Cardiology Context |
| Mean LDL-C | ~272 mg/dL (mean) | Classified as very high risk; exceeds typical FH diagnostic threshold (~190 mg/dL). |
| 1-Year NCPV Change | +42% median increase | Substantially higher than low-risk reference cohorts and comparable high-risk groups including those with FH or diabetes. |
| Zero CAC at Baseline | 57% of participants | Suggests initial resilience; however, absence of calcified plaque does not exclude non-calcified plaque burden. |
| ApoB Prediction of Progression | No statistically significant association | Conflicts with population-level dose-response data from Mendelian randomization and meta-analyses; may reflect insufficient power or follow-up duration. |
| Control Group | Absent | Prevents determination of whether observed progression rates are ‘modest’ or elevated relative to metabolically healthy individuals with low LDL-C. |
5.2 Critical Methodological Limitations
The interpretation offered by the KETO-CTA authors has attracted substantial criticism from the broader scientific community, culminating in an Expression of Concern issued by the Journal of the American College of Cardiology. Several critical methodological limitations undermine the authors’ conclusions.
The observation that ‘plaque begets plaque’ is a well-established phenomenon across all population groups and does not constitute evidence against the factors that initiated the plaque in the first place. The absolute rate of non-calcified plaque volume (NCPV) progression reported—approximately 42% over one year—is markedly higher than progression rates observed in high-risk groups such as those with diabetes or established FH in comparable longitudinal imaging studies.
The absence of a control group of lean, metabolically healthy individuals with low LDL-C makes it impossible to determine whether the progression observed was modest or alarming in relative terms. Without this comparator, the KETO-CTA data remains an intriguing but isolated observation that cannot be used to overturn decades of convergent causal evidence. The study’s one-year duration is also fundamentally insufficient to capture the decades-long kinetics of atherogenesis that Mendelian randomization studies reveal.
6. How ‘Sophisticated Doubt’ Functions in Scientific Discourse
‘Sophisticated doubt’ is a rhetorical technique that employs nuance, partial truths, and the amplification of scientific uncertainty to make established conclusions appear weaker than the totality of the evidence warrants. In the context of the LDL debate, this manifests in several recognizable patterns.
The ‘mass balance’ challenge: critics of the LEM argue that the model fails to account for where the additional cholesterol in LMHR LDL particles comes from if not from over-synthesis or under-clearance, an argument from basic lipid homeostasis that proponents address with mechanistic complexity but frequently without resolution at the level of first principles. Selective burden of proof: demanding a 10-year RCT in LMHR individuals—which would be ethically untenable if substantial cardiovascular risk is presumed—while accepting a one-year observational imaging study as definitive evidence of safety. Confounding relative and absolute risk: invoking the strong negative predictive value of a zero CAC score to imply that the relative atherogenic risk of extreme hyperlipidemia is negligible.
By framing the debate as a conflict between ‘individualized medicine’ and ‘population-level dogma,’ communicators tap into the contemporary zeitgeist of patient autonomy and institutional distrust—a rhetorically powerful but epistemologically incomplete position.
7. How Scientific Consensus Forms
It is a common misconception that scientific consensus is produced by a single definitive experiment. In reality, consensus emerges through the accumulation of ‘epistemic resilience’ via multiple independent processes. First, replication: the same association must be observed in different populations, by independent researchers, using methodologically distinct approaches. Second, convergence of methods: when genetic studies (nature’s experiments), epidemiological cohorts (observational data), and RCTs (interventional data) converge on the same directional finding, the probability of systematic error across all domains simultaneously approaches a very low threshold. Third, predictive success: a causal model must not only explain existing data but successfully predict the outcomes of novel interventions. The consistent reduction in cardiovascular events across hundreds of trials using diverse LDL-lowering mechanisms constitutes powerful prospective validation of the causal model.
In contrast, doubt is often constructed rhetorically by identifying a single anomaly—such as the elderly paradox or the LMHR phenotype—and using it to question the entire causal foundation. In science, an anomaly is an invitation to refine a model (for example: ‘LDL is causal, but its impact is modified by baseline insulin sensitivity and inflammatory milieu’) rather than to discard it wholesale. The two positions are not equivalent; one expands scientific understanding while the other selectively dismantles it.
8. Communication Strategies for Engaging the Skeptical Thinker
Effective clinical communication with LDL-skeptical patients requires moving beyond the ‘deficit model’—the assumption that providing more facts will resolve disagreement—toward an engagement model built on shared goals and collaborative reasoning.
8.1 Leading with Shared Goals
Rather than leading with a corrective posture, begin by affirming the shared objective of long-term health optimization. For example: ‘The metabolic improvements you’ve achieved on a carbohydrate-restricted diet are genuinely impressive and clinically meaningful. My concern is whether we can preserve those gains while also addressing the long-term implications of sustained, extreme elevations in circulating ApoB-containing particles.’ This framing positions the clinician as a collaborator rather than an adversary.
8.2 The Truth Sandwich and Prebunking
When addressing a specific claim, the ‘truth sandwich’ structure is effective: lead with the established fact, briefly address the misconception, and return to the established fact. For example: LDL particles play vital physiological roles including contributions to innate immunity and lipid transport. However, the claim that these functions render LDL categorically non-atherogenic is not supported by the mechanistic or outcomes evidence. The same particles that serve these functions, when present in excess over decades, become entrapped in the arterial intima—the necessary initiating event of atherosclerosis.
Prebunking involves inoculating the patient against specific misleading rhetorical tactics in advance: ‘You may encounter studies showing no correlation between LDL and plaque progression over a single year. Atherosclerosis is a decades-long process; one-year imaging snapshots, particularly without a control group, provide insufficient resolution to assess lifetime cumulative risk.’
8.3 Motivational Interviewing Example
Patient: “I’ve reviewed Dr. Norwitz’s data, and my metabolic markers are excellent. I don’t believe my elevated LDL is a meaningful risk factor in my specific case.”
Clinician (Reflective Listening): “It sounds like you’ve done considerable research and have concluded that your excellent metabolic health substantially attenuates the usual risks associated with elevated LDL-C. Can you tell me more about what evidence would change your assessment?”
Clinician (Open-Ended Question): “If we were to find evidence—using imaging available now—that plaque is already accumulating in your coronaries at an accelerated rate despite your metabolic health, how would that change your thinking about your current approach?”
9. Historical Analogies and Their Limitations
LDL skeptics sometimes compare their position to historical instances of correct scientific dissent—such as Warren and Marshall’s discovery of H. pylori as the cause of peptic ulcer disease in opposition to the prevailing ‘acid paradigm.’ This analogy is instructive but ultimately inapposite: the H. pylori hypothesis was initially opposed by a single established paradigm and was validated by an immediate, verifiable therapeutic response. LDL causality, by contrast, is supported by convergent evidence from genetics, pathophysiology, epidemiology, and interventional trials—a qualitatively different evidentiary structure that is far more resistant to overturning by a single anomalous finding.
A more instructive analogy is the dose-response relationship between cigarette smoking and lung cancer. Not every smoker develops cancer, and some non-smokers do—reflecting individual variability and the multifactorial nature of carcinogenesis. However, the causal role of smoking is undeniable because risk is a function of cumulative exposure: more pack-years predicts greater risk. Similarly, the LMHR individual claiming safety from atherosclerosis based on excellent metabolic health is analogous to a long-term smoker claiming safety based on superior pulmonary function and cardiovascular fitness. The fitness modifies the risk; it does not remove the causal exposure.
10. Conclusion
The most intellectually honest position currently available is one of cautious metabolic respect. We must respect the profound improvements in metabolic health that many individuals achieve through carbohydrate restriction, and we must equally respect the overwhelming convergent evidence that elevated ApoB-containing lipoproteins are the primary drivers of atherosclerosis regardless of the metabolic context that generates the elevation.
The Lean Mass Hyper-Responder phenotype represents a fascinating natural experiment—one that warrants rigorous, well-controlled longitudinal investigation. The KETO-CTA study, rather than closing the debate, has highlighted the urgent need for prospective, controlled, long-duration research in this population. For individuals currently presenting with this phenotype, the most prudent approach is individualized risk assessment using advanced vascular imaging, with explicit acknowledgment that ‘excellent metabolic health’ has not yet been demonstrated to negate the long-term consequences of extreme, sustained hyperlipidemia.
10.1 Questions Skeptics Are Right to Ask
- To what quantitative degree does a near-zero inflammatory environment (hs-CRP <0.3 mg/L) attenuate the atherogenicity of a given ApoB particle concentration?
- Why do some individuals with LDL-C >300 mg/dL remain free of detectable plaque into their eighth decade of life?
- Is there a saturation threshold for proteoglycan binding beyond which higher LDL concentrations no longer increase retention in a linear fashion?
- Can functional biomarkers of LDL retention be developed that provide superior individualized risk stratification compared to circulating LDL-C or ApoB concentration alone?
10.2 What the Totality of Evidence Still Supports
- The subendothelial retention of ApoB-containing lipoproteins is the necessary initiating event in atherosclerosis.
- Lifetime cumulative LDL-C exposure (‘cholesterol-years’) is a superior predictor of atherosclerotic risk compared to any single-point measurement.
- Genetic variants that lower LDL-C from birth confer profound protection against ASCVD, independent of other lifestyle factors.
- Lowering LDL-C and ApoB via any mechanism—diet, statins, ezetimibe, or PCSK9 inhibitors—consistently reduces major cardiovascular events across the full spectrum of baseline risk.
- Metabolic health modifies the rate and clinical impact of plaque progression but has not been demonstrated to negate the fundamental causal role of LDL in the atherosclerotic process.
References
The following references are limited to peer-reviewed journal articles and evidence-based scientific sources. Non-peer-reviewed sources including blog posts, podcasts, and popular media that appeared in earlier drafts have been removed from this reference list.
- Nordestgaard BG, Chapman MJ, Humphries SE, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease. Eur Heart J. 2013;34(45):3478-3490. doi:10.1093/eurheartj/eht273
- Ference BA, Ginsberg HN, Graham I, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2017;38(32):2459-2472. doi:10.1093/eurheartj/ehx144
- Tabas I, Williams KJ, Boren J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 2007;116(16):1832-1844. doi:10.1161/CIRCULATIONAHA.106.676890
- Ference BA, Bhatt DL, Catapano AL, et al. Association of genetic variants related to combined exposure to lower low-density lipoproteins and lower systolic blood pressure with lifetime risk of cardiovascular disease. JAMA. 2019;322(14):1381-1391. doi:10.1001/jama.2019.14120
- Rawlings AM, Sharrett AR, Schneider ALC, et al. Diabetes in midlife and cognitive change over 20 years: a cohort study. Ann Intern Med. 2014;161(11):785-793.
- Norwitz NG, Feldman D, Soto-Mota A, Kalayjian T, Ludwig DS. Elevated LDL cholesterol with a carbohydrate-restricted diet: evidence for a ‘lean mass hyper-responder’ phenotype. Curr Dev Nutr. 2022;6(1):nzab144. doi:10.1093/cdn/nzab144
- Norwitz NG, Soto-Mota A, Feldman D, Budoff M, Rapaport S. The Lipid Energy Model: reimagining lipoprotein function in the context of carbohydrate-restricted diets. Metabolites. 2022;12(5):460. doi:10.3390/metabo12050460
- Lawler PR, Akinkuolie AO, Ridker PM, et al. Discordance between circulating atherogenic cholesterol mass and lipoprotein particle concentration in relation to future coronary events in women. Clin Chem. 2017;63(1):314-323. doi:10.1373/clinchem.2016.263566
- Ravnskov U, Diamond DM, Hama R, et al. Lack of an association or an inverse association between low-density-lipoprotein cholesterol and mortality in the elderly: a systematic review. BMJ Open. 2016;6(6):e010401. doi:10.1136/bmjopen-2015-010401
- Baigent C, Blackwell L, Emberson J, et al; Cholesterol Treatment Trialists’ (CTT) Collaboration. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376(9753):1670-1681. doi:10.1016/S0140-6736(10)61350-5
- Silverman MG, Ference BA, Im K, et al. Association between lowering LDL-C and cardiovascular risk reduction among different therapeutic interventions: a systematic review and meta-analysis. JAMA. 2016;316(12):1289-1297. doi:10.1001/jama.2016.13985
- Sabatine MS, Giugliano RP, Keech AC, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376(18):1713-1722. doi:10.1056/NEJMoa1615664
- Soto-Mota A, Norwitz NG, Evans RD, Clarke K, Barber TM. Cholesterol is not created equal: the effect of the Oreo cookie experiment in context. Curr Opin Endocrinol Diabetes Obes. 2023;30(2):128-136. doi:10.1097/MED.0000000000000786
- Budoff MJ, Mayrhofer T, Ferencik M, et al. Prognostic value of coronary artery calcium in the PROMISE study (Prospective Multicenter Imaging Study for Evaluation of Chest Pain). Circulation. 2017;136(21):1993-2005. doi:10.1161/CIRCULATIONAHA.117.030578
- Soto-Mota A, Norwitz NG, Budoff M, et al. Plaque begets plaque, ApoB does not: longitudinal data from the KETO-CTA trial. JACC Adv. 2025;4(5):101480. doi:10.1016/j.jacadv.2025.101480 [Expression of Concern issued by the Journal]
- Williams KJ, Tabas I. The response-to-retention hypothesis of atherogenesis reinforced. Curr Opin Lipidol. 1998;9(5):471-474. doi:10.1097/00041433-199810000-00012
- Ference BA, Yoo W, Alesh I, et al. Effect of long-term exposure to lower low-density lipoprotein cholesterol beginning early in life on the risk of coronary heart disease: a Mendelian randomization analysis. J Am Coll Cardiol. 2012;60(25):2631-2639. doi:10.1016/j.jacc.2012.09.017
- Grundy SM, Stone NJ, Bailey AL, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol. J Am Coll Cardiol. 2019;73(24):e285-e350. doi:10.1016/j.jacc.2018.11.003
- Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;23(R1):R89-R98. doi:10.1093/hmg/ddu328
- Boren J, Williams KJ. The central role of arterial retention of cholesterol-rich apolipoprotein-B-containing lipoproteins in the pathogenesis of atherosclerosis: a triumph of simplicity. Curr Opin Lipidol. 2016;27(5):473-483. doi:10.1097/MOL.0000000000000330
- Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377(12):1119-1131. doi:10.1056/NEJMoa1707914
- Khera AV, Kathiresan S. Genetics of coronary artery disease: discovery, biology and clinical translation. Nat Rev Genet. 2017;18(6):331-344. doi:10.1038/nrg.2016.160
- Catapano AL, Pirillo A, Bonacina F, Norata GD. HDL in innate and adaptive immunity. Cardiovasc Res. 2014;103(3):372-383. doi:10.1093/cvr/cvu150
- Swerdlow DI, Preiss D, Kuchenbaecker KB, et al; DIAGRAM Consortium; MAGIC Consortium; InterAct Consortium. HMG-coenzyme A reductase inhibition, type 2 diabetes, and bodyweight: evidence from genetic analysis and randomised trials. Lancet. 2015;385(9965):351-361. doi:10.1016/S0140-6736(14)61183-1
- Toth PP, Larsen WE, Zhu X, et al. Causal relationship between drug target genes of LDL-cholesterol lowering pharmacotherapies and cardiovascular disease: a drug target Mendelian randomization study. Front Cardiovasc Med. 2025;12:1484521. doi:10.3389/fcvm.2025.1484521