
Early Lipid-Lowering Therapy Across the Lifespan
Optimizing Lifetime Apolipoprotein B Exposure to Prevent Atherosclerotic Cardiovascular Disease
Abstract
Atherosclerotic cardiovascular disease (ASCVD) remains the leading cause of death worldwide, accounting for an estimated 19.8 million deaths in 2022. 1, 2 A convergent body of pathophysiological, epidemiological, genetic, and randomized evidence indicates that apolipoprotein B (ApoB)-containing lipoproteins are regarded, under the response-to-retention framework and current genetic evidence, as the necessary initiating agents of atherogenesis: the disease begins when these particles are retained within the arterial intima, provoking a chronic maladaptive inflammatory response that accumulates silently over decades. 3, 4, 12 Because arterial injury is a cumulative product of both the concentration of atherogenic particles and the duration of exposure—quantified for LDL cholesterol as cumulative “LDL-C-years” and extended conceptually to the particle count as “ApoB-years”—the timing of intervention is as consequential as its intensity. Mendelian-randomization studies suggest that lifelong genetically mediated exposure to lower LDL-C is associated with substantially greater coronary protection per unit difference than that observed during the shorter follow-up of trials in which pharmacologic therapy is initiated later in life. 13, 14 This narrative review synthesizes the primary literature underlying that argument across the lifespan; evaluates the quantitative evidence for lifestyle and pharmacologic ApoB reduction; details the comparative pharmacology of statin and non-statin agents; and weighs the adverse-effect profile of lipid-lowering therapy. The available evidence supports earlier consideration of sustained ApoB lowering, with treatment intensity individualized according to cumulative exposure, absolute ASCVD risk, comorbidities, patient preferences, and the balance of expected benefit and harm.
This article is a narrative review of mechanistic, genetic, randomized-trial, and guideline evidence on cumulative ApoB exposure and cardiovascular risk. It is not a systematic review or meta-analysis and does not include formal risk-of-bias scoring; its conclusions should be interpreted in light of possible selection and emphasis bias inherent to the narrative format. 53
Keywords: apolipoprotein B; LDL cholesterol; atherosclerosis; response-to-retention; cumulative exposure; Mendelian randomization; primary prevention; statins; PCSK9 inhibition; inclisiran; lipoprotein(a).
Methods and Scope
This is a narrative review; the account below is provided for transparency rather than as a claim to systematic-review methodology. 53, 54 The literature underpinning each section was identified through targeted searching of PubMed/MEDLINE and the Cochrane Library, supplemented by the reference lists of major society guidelines and by hand-searching of landmark trials, for records available through early 2026. Priority was given to primary sources—randomized controlled trials, Mendelian randomization studies, individual-patient-data and study-level meta-analyses, and official guideline documents—over reviews and tertiary sources; where a primary source was not accessible in full, peer-reviewed abstracts or the official trial report were used. Quantitative values reported in the summary tables were extracted from the cited primary trials or guideline documents and are footnoted to those sources at the level of the individual estimate; qualitative comparative categories (for example, relative muscle-symptom liability) are explicitly labeled as interpretive syntheses rather than validated ranking scales. No formal systematic-review reporting protocol (e.g., PRISMA 2020, a reporting guideline for systematic reviews) or structured review-appraisal instrument (e.g., AMSTAR 2 or ROBIS, which assess the quality of systematic reviews) was applied, and no formal risk-of-bias scoring of individual primary studies was undertaken; the selection of evidence necessarily reflects editorial judgment. This is a recognized limitation of the narrative format and is revisited in the Limitations section. 53, 54
1. Biological Basis of Apolipoprotein B–Containing Lipoprotein Causality
Atherosclerotic cardiovascular disease is the leading cause of global mortality. 1, 2 Decades of pathophysiological, epidemiological, and genetic research have established that apolipoprotein B (ApoB)-containing lipoproteins are regarded as the necessary initiating factor in atherogenesis under the currently accepted response-to-retention framework: they are the agents whose subendothelial retention sets the disease in motion, even as numerous other biological processes—endothelial dysfunction, disturbed flow, hypertension, hyperglycemia, oxidative stress, and inflammatory signaling—influence how the resulting plaque progresses once retention has occurred. 3, 12, 55 The initiation of atherosclerosis is governed by the response-to-retention hypothesis, first formalized by Williams and Tabas and subsequently extended by contemporary work on lipoprotein retention and modification. 3, 4 According to this framework, the key initiating event is the subendothelial retention of ApoB-containing lipoproteins within the tunica intima of susceptible arterial segments. 3, 4 Critically, the original formulation demonstrated that predisposing stimuli—hemodynamic, inflammatory, or metabolic—in the absence of an abundant supply of atherogenic lipoproteins are insufficient to initiate the disease. Retention is the non-redundant first step, without which the downstream processes do not produce atheroma; it is not, however, the only determinant of the disease that follows. 3, 55
To enter the subendothelial space, circulating lipoproteins must cross the vascular endothelial barrier. Although receptor-mediated transcytosis appears to predominate under physiological conditions, the relative contribution of different transport routes in human atherosclerosis remains under active investigation, and passive paracellular entry through regions of endothelial dysfunction, disturbed flow, or injury probably contributes under pathological conditions. 4, 5, 6 Experimental studies identify scavenger receptor B1 (SR-B1)- and activin receptor-like kinase 1 (ALK1)-dependent pathways as important mechanisms of LDL transcytosis across intact endothelium. 5, 6 ALK1 in particular mediates an LDL-receptor–independent route of entry, which helps explain why lowering the circulating particle number is so effective at limiting intimal delivery: clearance pathways can be bypassed, and higher circulating particle concentrations are expected to increase opportunities for arterial-wall entry and retention. 5, 6 This process allows lipoproteins across the size range of LDL and smaller remnant particles to access the intima.
Once in the subendothelial space, these lipoproteins interact with the extracellular matrix. 4 The ApoB-100 molecule on the lipoprotein surface contains basic, positively charged amino-acid domains. 3 These domains bind ionically to the negatively charged glycosaminoglycan (GAG) side chains of arterial proteoglycans—such as chondroitin sulfate and heparan sulfate—which are synthesized by vascular smooth-muscle cells (SMCs). 3, 4 This electrostatic entrapment prevents the lipoproteins from diffusing back into the circulation and is the physical event that converts a transient exposure into a durable, disease-initiating deposit. 3
Retained particles undergo chemical modifications, including oxidation by local free radicals and enzymatic cleavage by secretory phospholipase A2 (sPLA2) and sphingomyelinase. 4, 7 These modifications promote aggregation and fusion of the retained lipoproteins, which in turn triggers a chronic, maladaptive inflammatory response. 4 The endothelium becomes activated and expresses cell-adhesion molecules that recruit circulating monocytes into the intima. Monocytes differentiate into macrophages, which internalize the modified, aggregated lipoproteins via scavenger receptors. Because these receptors—unlike the LDL receptor—are not subject to sterol feedback inhibition, macrophages accumulate cholesteryl esters without restraint and transform into lipid-laden foam cells. 4 Over decades, foam-cell accumulation, SMC migration, and extracellular-matrix deposition drive plaque progression and necrotic-core formation. 4 The inflammatory response is therefore largely downstream of, and dependent upon, the retained ApoB particle; it is the principal mechanism by which retention becomes disease. This causal ordering is why the residual inflammatory risk that persists after aggressive lipid lowering—real, measurable, and independently treatable—does not displace the particle from its position as the primary initiating target, even as it identifies a complementary one (Section 6.6). 52
1.1 Distinguishing the Standard Lipid Biomarkers
Understanding this biological cascade requires distinguishing between the standard lipid biomarkers, which are not interchangeable and which can diverge substantially in clinically important ways:
- Low-Density Lipoprotein Cholesterol (LDL-C). This measures the mass of cholesterol carried within LDL particles per unit volume of plasma. 8 While it serves as the traditional surrogate for cardiovascular risk, LDL-C reflects neither particle concentration nor structural heterogeneity, and it may underestimate atherogenic particle burden when LDL particles are relatively cholesterol-depleted. 8
- Apolipoprotein B (ApoB). Each atherogenic particle—LDL, VLDL, IDL, and TRL remnants (each carrying one ApoB-100), and lipoprotein(a)—contains a single ApoB molecule, so plasma ApoB concentration is a near-direct stoichiometric count of circulating atherogenic particles. 8 (Intestinally-derived chylomicrons and their remnants carry the truncated ApoB-48 isoform; standard clinical ApoB assays measure predominantly ApoB-100 and, in the fasting state, chylomicron contribution is negligible.) Approximately 90% of circulating ApoB resides on LDL particles, and ApoB captures the physical exposure of the arterial wall to plaque-initiating agents more faithfully than a cholesterol-mass measurement. 8
- Non-High-Density Lipoprotein Cholesterol (Non-HDL-C). Calculated as total cholesterol minus HDL-C, this parameter quantifies the cumulative cholesterol mass within all atherogenic lipoproteins. 8, 51 It serves as a close surrogate for atherogenic particle mass—capturing remnant cholesterol—but remains subject to compositional bias. 8
- Remnant Cholesterol. This represents the cholesterol content of triglyceride-rich lipoproteins (TRLs), specifically VLDL and IDL particles. 4 These remnants are highly atherogenic and can access the subendothelium directly. 4
- Lipoprotein(a) [Lp(a)]. A genetically determined, highly atherogenic variant in which an LDL-like, ApoB-containing particle is covalently bound to apolipoprotein(a). 8 Lp(a) contributes to both plaque progression and localized thrombosis, and it is essentially unmodifiable by lifestyle. 8, 46
1.2 Genetic Evidence Relating ApoB Particle Number to Coronary Risk
Mendelian randomization (MR) studies—which exploit the random allocation of genetic variants at conception to approximate a lifelong randomized experiment—provide strong genetic evidence that ApoB is the principal causal lipid driver of atherosclerosis. 9, 12 When discordance exists between LDL-C and ApoB—commonly in insulin resistance, obesity, type 2 diabetes, or hypertriglyceridemia—cardiovascular risk tracks with ApoB rather than with LDL-C. 8 In these states the liver overproduces triglyceride-rich VLDL, yielding not only a high number of small, dense LDL particles but also an excess of atherogenic remnant lipoproteins; standard LDL-C measurement then underestimates the true atherogenic particle concentration, whereas ApoB counts every atherogenic particle regardless of its cholesterol content. 8 Framed precisely, risk is caused by the biology of particle retention rather than by any measurement of it: ApoB is best understood not as the cause of risk but as the strongest validated circulating biomarker of the causal burden of atherogenic lipoproteins. 8, 12
The most direct evidence comes from multivariable MR. When LDL-C, triglycerides, and ApoB are analyzed jointly, the associations of LDL-C and triglycerides with coronary artery disease become null once ApoB is accounted for, whereas ApoB retains a strong, independent causal association. In the landmark analysis of lipoprotein lipase (LPL) and LDL-receptor (LDLR) variants, after conditioning on ApoB the odds ratio for coronary heart disease was 1.01 per standard deviation for both LDL-C and triglycerides (both non-significant), while ApoB retained a strong independent association—an odds ratio of 0.76 per standard-deviation lower ApoB (i.e., a 24% lower risk of coronary heart disease per SD reduction in ApoB; P = 7.5 × 10⁻²⁰). 9 Independent high-throughput and multivariable MR analyses reproduce this ordering, consistently prioritizing ApoB as the principal lipid-related causal factor for coronary artery disease. 10, 11 The clinical corollary is that where LDL-C and ApoB diverge, risk often tracks more closely with the particle count; in such settings ApoB should materially inform risk estimation and treatment decisions alongside LDL-C, non-HDL-C, overall ASCVD risk, and guideline context, rather than serving as a sole automatic determinant. 8, 12, 55 The incremental value of measuring ApoB is greatest precisely when it is discordant with LDL-C—when the cholesterol content of the particles misrepresents their number—because it is in exactly those patients that LDL-C-guided assessment most often under-recognizes the true atherogenic burden. 8
2. The Cumulative Lifetime Burden: “ApoB-Years”
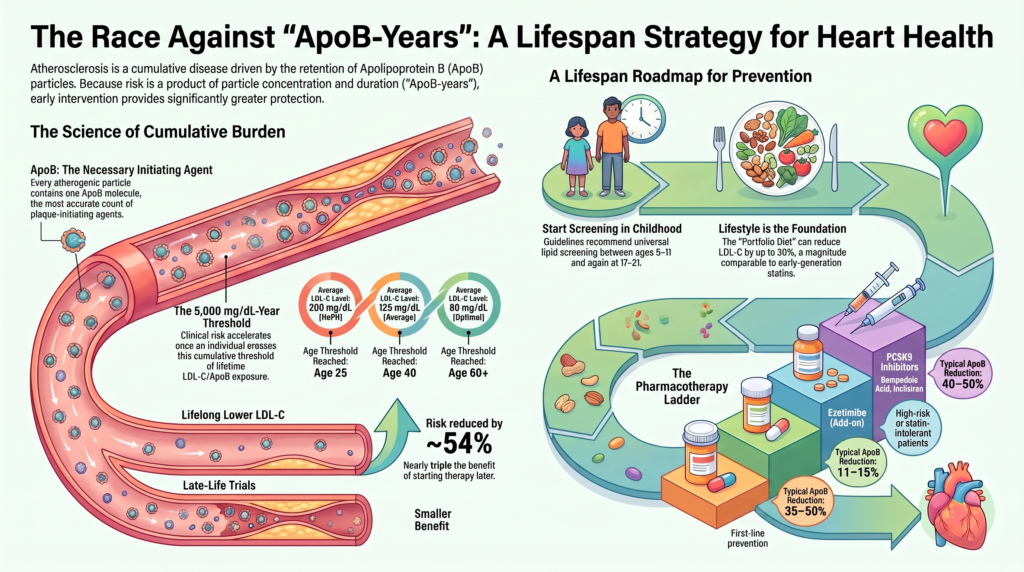
The clinical progression of ASCVD is determined by both the concentration of circulating atherogenic particles and the duration of arterial-wall exposure to them. 8, 13 This lifetime burden is captured by the concept of cumulative exposure—most rigorously quantified for LDL cholesterol as “LDL-C-years” (the product of average LDL-C and years of exposure), and extended on biological grounds to the particle count as “ApoB-years.” 13, 14 The specific quantitative thresholds cited below derive from the LDL-C cumulative-exposure model of Ference, Braunwald, and Catapano; the ApoB-years formulation is a mechanistically-motivated conceptual extension rather than a separately validated quantitative threshold, and it has not been calibrated as an individual-level clinical calculator. 13 Atherosclerosis progresses silently for decades, and clinical events typically occur only once a critical cumulative threshold has been crossed. 13
The cumulative-exposure framework formalized by Ference, Braunwald, and Catapano proposes that the incidence of clinical coronary events begins to rise more substantially after approximately 5,000 mg/dL-years of cumulative LDL-C exposure. This value is a model-derived, population-level heuristic rather than a universal biological threshold: individual events may occur at substantially lower or higher cumulative exposure depending on smoking, blood pressure, diabetes, Lp(a), sex, genetic background, inflammatory burden, and plaque morphology. 13 The consequences of this arithmetic are best illustrated by contrasting exposure trajectories, presented here as model-derived projections from the cumulative-exposure hypothesis rather than as directly measured incidence data:
- For an individual whose lifetime average LDL-C is approximately 125 mg/dL, the ~5,000 mg/dL-year threshold is reached near age 40. In the model, the cumulative incidence of myocardial infarction then rises in an accelerating fashion with each subsequent decade of continued exposure, roughly doubling per decade as cumulative exposure climbs from ~5,000 toward ~10,000 mg/dL-years between ages 40 and 80. 13
- A person with heterozygous familial hypercholesterolemia (HeFH) and a lifetime average LDL-C near 200 mg/dL crosses the same threshold roughly two decades earlier—around age 25—which accounts for the premature ASCVD that defines the condition. 13, 20
- Conversely, an individual maintaining a lifetime average LDL-C near 80 mg/dL does not cross the threshold until the seventh decade of life. 13, 14
The direction and magnitude of these projections are corroborated by cohort data linking early-adulthood lipid exposure to later events, even after adjustment for later-life lipid levels. 22, 23
2.1 Short-Term Trials Versus Lifelong Genetic Evidence
The biological benefit of early lipid lowering is demonstrated by comparing the outcomes of short-term randomized controlled trials (RCTs) with the lifelong protection shown in Mendelian randomization studies. 13, 14
- Short-term (5-year RCTs). Meta-analyses from the Cholesterol Treatment Trialists’ (CTT) Collaboration show that each 1.0 mmol/L (38.7 mg/dL) reduction in LDL-C reduces the relative risk of major vascular events by approximately 22% over roughly five years, with the absolute risk reduction and number needed to treat depending on the patient’s baseline risk. 15, 16
- Long-term (lifelong MR). In the pivotal analysis of long-term exposure beginning early in life, each 1 mmol/L lower LDL-C sustained from birth was associated with a 54.5% (95% CI 48.8–59.5%) lower risk of coronary heart disease—roughly a threefold greater reduction per unit than that achieved with a statin started later in life (P = 8.4 × 10⁻¹⁹). 14, 13
The lifespan consequences are commensurate with the event data. Genetic analysis indicates that each ~1 standard deviation (≈ 38 mg/dL) higher LDL-C is associated with approximately 1.2 fewer years of life (95% CI −1.55 to −0.87) and about 28% lower odds of surviving to the 90th percentile of age. 17 This gap between short-term and lifelong benefit is not a paradox; it is the signature of a cumulative disease. Late-initiated therapy stabilizes plaque that already exists but leaves a large residual burden of established, rupture-prone lesions. Earlier intervention may delay lesion formation and reduce cumulative exposure to atherogenic particles, potentially keeping cumulative LDL-C exposure below the modelled range associated with accelerating clinical risk. 13, 14 The logical consequence is that the greatest lifetime protection is obtained by lowering ApoB earlier and keeping it low—not by lowering it aggressively only after risk has already declared itself.
3. Clinical Initiation Thresholds and Screening Across the Lifespan
Cardiovascular risk assessment has shifted toward a life-course prevention framework, employing distinct lipid-lowering strategies across different age groups and calibrated to absolute risk. 18
3.1 Children and Adolescents (Ages 2–17 Years)
Pediatric lipid guidelines recommend universal screening with a non-fasting lipid panel once between the ages of 9 and 11 years, and again between 17 and 21 years. 19 Universal screening at 9–11 years is timed to identify HeFH before puberty, as LDL-C levels can temporarily decline by 10–20% during pubertal development, which reduces the sensitivity of screening performed mid-puberty. 19 Selective screening using a fasting lipid panel is recommended starting at age 2 years for children with a family history of premature ASCVD (before age 55 in male relatives, before age 65 in female relatives) or severe parental hypercholesterolemia. 19
Guidelines for pediatric HeFH management differ across clinical organizations:
- European Atherosclerosis Society (EAS). Pharmacologic treatment for children with confirmed HeFH is generally initiated from age 8–10 years. For children aged 8–10 years, the aim is a ≥ 50% reduction from the pre-treatment LDL-C; from age 10 years onward, the target is an LDL-C < 3.5 mmol/L (130 mg/dL). 20
- American Academy of Pediatrics (AAP) & National Lipid Association (NLA). Statin therapy is generally initiated from age 10 years (occasionally 8–10) in children with an LDL-C ≥ 190 mg/dL despite a 6- to 12-month trial of lifestyle changes, or an LDL-C ≥ 160 mg/dL in the presence of a family history of premature CVD or additional risk factors. The target is a ≥ 50% reduction in LDL-C or an absolute level < 130 mg/dL. 19
For homozygous familial hypercholesterolemia (HoFH), early diagnosis and intensive treatment are required in infancy, with the goal of keeping LDL-C < 3.0 mmol/L (115 mg/dL), or < 2.5 mmol/L (100 mg/dL) if clinical ASCVD is established. 21 Across all pediatric scenarios, pharmacologic treatment decisions should generally involve clinicians experienced in pediatric lipid disorders and proceed through shared decision-making with the child’s family, balancing the long-term rationale for early lowering against the practical and psychosocial considerations of initiating lifelong therapy in childhood. 19, 20
3.2 Young Adults (Ages 18–39 Years)
Data from the CARDIA study show that elevated ApoB and LDL-C exposure in young adulthood is associated with a higher risk of ASCVD events and subclinical atherosclerosis after age 40, independent of later lipid levels. 22, 23 A single lipid panel obtained in young adulthood can predict decades of cumulative exposure. 22 For primary prevention in young adults with an LDL-C ≥ 160 mg/dL (≈ 4.1 mmol/L) or an ApoB ≥ 130 mg/dL together with a family history of premature ASCVD, initiating statin therapy should be considered to prevent the early accumulation of ApoB-years. 18, 22
3.3 Adults (Ages 40–75 Years)
The 2026 ACC/AHA/Multisociety Dyslipidemia Guideline recommends using the PREVENT equations to estimate 10-year ASCVD risk. 18 This tool categorizes risk into graded strata:
- Low Risk (< 3%): The focus is on healthy lifestyle habits. 18
- Borderline Risk (3% to < 5%): At borderline estimated risk, the absolute cardiovascular benefit of statin therapy is modest and depends on the accuracy of the risk estimate, the magnitude of LDL-C reduction, treatment duration, and the presence of risk-enhancing factors. 16 Statins also produce a small increase in incident diabetes, concentrated predominantly among patients with pre-existing metabolic susceptibility. 37 Because both the benefits and the harms vary considerably across individuals—and because a prevented cardiovascular event and an incident case of diabetes are not clinically equivalent outcomes—decisions in this range should emphasize shared decision-making informed by risk-enhancing factors and patient priorities rather than a universal treatment rule. 16, 18
- Intermediate Risk (5% to 10%): Statin initiation is recommended, targeting a ≥ 30–50% reduction in LDL-C. 18
- High Risk (> 10%): Statin initiation is strongly recommended, targeting a ≥ 50% reduction in LDL-C. 18
The 2026 guidelines restore absolute LDL-C and non-HDL-C treatment goals based on baseline risk:
- Secondary Prevention (Established ASCVD): Target LDL-C < 55 mg/dL (and non-HDL-C < 85 mg/dL) for very-high-risk patients, and < 70 mg/dL for other patients with established ASCVD. 18
- Primary Prevention with elevated baseline LDL-C: Goals stratified by baseline LDL-C, with a lower target (e.g., < 100 mg/dL, or lower) where HeFH, additional risk factors, or subclinical atherosclerosis is present. 18
- Subclinical Atherosclerosis (Coronary Artery Calcium): The guideline expands CAC for reclassification—any CAC > 0 supports initiating therapy, and CAC ≥ 100 Agatston units or ≥ the 75th percentile supports high-intensity lowering, while a CAC of 0 in the absence of high-risk features may permit deferral with reassessment. Specific absolute LDL-C targets keyed to each CAC band should be read from the current guideline tables. 18
The Canadian Cardiovascular Society (CCS) recommends screening all individuals ≥ 40 years of age (or earlier in high-risk ethnic groups), and preferring non-HDL-C or ApoB as the lipid marker when triglycerides are > 1.5 mmol/L (133 mg/dL). 24 In intermediate-risk individuals, CCS guidelines recommend initiating statin therapy when LDL-C ≥ 3.5 mmol/L, non-HDL-C ≥ 4.2 mmol/L, or ApoB ≥ 1.05 g/L. 24
3.4 Older Adults (Over Age 75 Years)
Clinical trial data support continuing statin therapy in older adults who are tolerating treatment. 16 For primary prevention in patients over 75 years of age, initiation is recommended after a clinician–patient discussion that weighs the expected cardiovascular benefit against potential drug–drug interactions, polypharmacy, and muscular tolerability. 16, 18
3.5 Special Triggers for Initiation
- Elevated ApoB with Normal LDL-C. This discordant phenotype generally reflects a greater number of cholesterol-depleted ApoB-containing particles, often including small dense LDL and triglyceride-rich remnant particles. In patients with type 2 diabetes, metabolic syndrome, or hypertriglyceridemia, an ApoB ≥ 130 mg/dL is a risk-enhancing factor that may support consideration of initiation or intensification of lipid-lowering therapy in appropriate clinical context—particularly when LDL-C is discordantly reassuring, triglycerides are elevated, or overall risk is uncertain—rather than serving as an automatic stand-alone trigger. 8, 18
- Elevated Lp(a). Universal screening of adults for Lp(a) is recommended at least once. 18 An Lp(a) ≥ 125 nmol/L (≈ 50 mg/dL) increases ASCVD risk approximately 1.4-fold, and an Lp(a) ≥ 250 nmol/L (≈ 100 mg/dL) increases risk approximately 2-fold. Elevated Lp(a) warrants more intensive risk-factor management and a lower target for LDL-C. 18, 46
4. Lifestyle Therapy and Primordial Prevention
Lifestyle change is a key component of dyslipidemia management and the foundation of primordial prevention, though its magnitude of effect is generally lower than that of pharmacotherapy. 18 The effects summarized below derive from randomized and meta-analytic data; where a range is given, the upper bound generally requires high adherence and substantial replacement of saturated fat.
- Mediterranean Diet. Emphasizes extra-virgin olive oil, vegetables, fruits, whole grains, legumes, and nuts, with moderate fish consumption and minimal red meat. Meta-analyses of randomized trials show that a Mediterranean pattern lowers LDL-C modestly relative to comparison diets—on the order of 5–10 mg/dL—and improves several cardiometabolic risk factors including endothelial function, insulin sensitivity, and markers of systemic inflammation; its cardiovascular benefits likely extend beyond lipid lowering alone. 62, 28
- Whole-Food Plant-Based Diets. Restricting animal products and emphasizing minimally processed plant foods lowers atherogenic lipoproteins: a meta-analysis of 30 randomized trials found that vegetarian and vegan diets reduced LDL-C by roughly 10% and ApoB by approximately 14% relative to omnivorous diets, with larger reductions under controlled feeding conditions. 63, 28
- Low Saturated Fat Diets. Restricting saturated fatty acids to < 7% of daily energy intake and replacing them with unsaturated fats upregulates hepatic LDL-receptor expression, increasing clearance of circulating ApoB particles and lowering LDL-C; in randomized dietary-replacement trials pooled by the American Heart Association, replacing saturated with polyunsaturated fat reduced cardiovascular events by roughly 30%, comparable to the effect of statin therapy. 28
- Soluble Viscous Fiber. Soluble fiber binds bile acids in the intestinal lumen, increasing their fecal excretion and depleting the hepatic cholesterol pool, which upregulates hepatic LDL receptors. 26 Dose-response data show a reduction in LDL-C on the order of ~1–2 mg/dL per gram per day of soluble fiber, so that ~10 g/day yields a clinically meaningful reduction and also lowers ApoB. 26
- Plant Sterols and Stanols. These structural analogs of cholesterol compete for incorporation into mixed micelles in the intestinal lumen, reducing cholesterol absorption. 27 A daily intake of 2–2.5 g of phytosterols is associated with an 8–10% reduction in circulating LDL-C and a significant accompanying reduction in ApoB. 27
- The Portfolio Diet. Combines plant sterols, viscous fibers, soy protein, and tree nuts. This diet reduces LDL-C by approximately 17% under free-living conditions and by up to ~30% under metabolically controlled conditions—a magnitude comparable to a first-generation statin—while also significantly reducing ApoB, triglycerides (by roughly 16%), and C-reactive protein. 25
- Aerobic and resistance training have a modest direct effect on LDL-C and ApoB concentrations but reduce cardiovascular risk through multiple lipid and non-lipid mechanisms, including improved cardiorespiratory fitness, insulin sensitivity, blood pressure, endothelial function, systemic inflammation, and autonomic regulation—so their modest lipid effect should not be mistaken for modest clinical importance. 29 Guidelines recommend a weekly target of ≥ 150 minutes of moderate-intensity or ≥ 75 minutes of vigorous-intensity aerobic exercise, supplemented by resistance training on at least 2 days. 29
- Other Behavioral and Metabolic Interventions. Several broader lifestyle exposures shape the ApoB trajectory indirectly. Weight loss reduces hepatic free-fatty-acid flux, lowering VLDL synthesis and circulating ApoB, and obesity is a central driver of the atherogenic dyslipidemia (high triglycerides, low HDL-C, small dense LDL) that produces LDL-C–ApoB discordance. High intake of ultra-processed foods is associated with adverse lipid profiles and higher cardiovascular risk, plausibly through displacement of fiber- and unsaturated-fat-rich whole foods and through excess refined carbohydrate and sodium. Poor sleep quality and short sleep duration are associated with dyslipidemia, insulin resistance, and hypertension, and excess alcohol raises triglycerides and blood pressure. Smoking cessation reduces endothelial dysfunction and oxidation of circulating ApoB particles, and blood-pressure control further protects the vascular wall. 18, 28 These exposures are not primarily ApoB-lowering interventions, but addressing them improves the metabolic milieu in which retained particles do their damage and is integral to primordial prevention. 18
While lifestyle modifications are effective for primary prevention in low-to-moderate-risk populations and are indispensable in all, they are rarely sufficient to meet target lipid levels in patients with familial hypercholesterolemia or established ASCVD. 19 In these clinical scenarios, delaying pharmacologic therapy unnecessarily increases cumulative ApoB exposure. 14
5. HMG-CoA Reductase Inhibitors: Comparative Pharmacokinetics and Tolerability
Statins are the first-line pharmacologic therapy for both primary and secondary cardiovascular prevention. 15 They act by competitively inhibiting 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase, the rate-limiting enzyme in hepatic cholesterol synthesis. 30, 31 This enzymatic blockade depletes the intrahepatic cholesterol pool, upregulating the expression of LDL receptors on the hepatocyte membrane, which increases the clearance of circulating ApoB-containing lipoproteins. 31 Statins are categorized as either lipophilic or hydrophilic based on their molecular substituents. 30
Lipophilic statins (atorvastatin, simvastatin, lovastatin, fluvastatin, and pitavastatin) have non-polar substituents, allowing them to translocate through cell membranes via passive diffusion. 30 They are more widely distributed throughout extrahepatic tissues, including skeletal muscle, which may increase the risk of muscular adverse effects. Pitavastatin is a notable case: although relatively lipophilic, it undergoes only minimal cytochrome-P450 metabolism, which favorably distinguishes its interaction profile. 30 Hydrophilic statins (rosuvastatin and pravastatin) contain polar substituents that restrict passive diffusion. 30, 31 Their uptake is highly selective for hepatocytes, mediated by carrier-mediated transport via organic anion-transporting polypeptide 1B1 (OATP1B1); this hepatoselectivity reduces passive entry into peripheral skeletal-muscle cells. 30, 31
5.1 Statin-Associated Muscle Symptoms in Proportion
The primary safety challenge to statin adherence is the development of muscle-related adverse events, and here the primary evidence demands precision rather than alarm. 32 These range from mild, subjective symptoms to rare, severe muscle injury:
- Statin-Associated Muscle Symptoms (SAMS). An umbrella term for muscle-related symptoms temporally associated with statin use, regardless of proven pharmacologic causality. In observational registries, SAMS are reported by roughly 10–15% (and up to ~29%) of statin users. However, in double-blind randomized controlled trials, the absolute difference in muscle symptoms between statin-treated and placebo-treated groups is small. 32 The SAMSON crossover trial is informative here, though its findings apply most directly to the population it studied—patients who had already discontinued a statin because of side effects. In that group, mean symptom intensity was 8.0 in months with no tablet, 15.4 on placebo, and 16.3 on atorvastatin—statistically indistinguishable from placebo—yielding a nocebo ratio of 0.90: roughly 90% of the reported symptom burden in these previously intolerant patients was reproduced by placebo. 32, 33 This does not mean the symptoms are imagined, nor should the 90% figure be generalized uncritically to all statin users; rather, in patients who report intolerance it reframes management away from permanent discontinuation and toward structured reassurance and rechallenge. The convergence of this crossover evidence with the small absolute excess seen in blinded trials nonetheless indicates that the drug accounts for only a minority of attributed muscle symptoms. 32
- Subjective muscle pain, aching, or stiffness in a symmetric distribution affecting large proximal muscle groups (thighs, buttocks, shoulders) in the absence of creatine kinase (CK) elevation. 32
- Muscle symptoms accompanied by biochemical evidence of muscle injury; commonly defined as a CK elevation greater than 10 times the upper limit of normal (ULN), although diagnostic thresholds vary across consensus definitions. 31
- A severe, potentially life-threatening form of myonecrosis characterized by profound muscle pain, weakness, dark urine, marked CK elevation, and acute kidney injury secondary to myoglobinuria. It is extremely uncommon with contemporary statin monotherapy at standard doses (on the order of ~0.44 hospitalizations per 10,000 person-years); risk rises substantially with high statin doses, major drug interactions, renal impairment, and concomitant gemfibrozil. 34
- Anti-HMGCR Immune-Mediated Necrotizing Myopathy (IMNM). A rare autoimmune myopathy characterized by progressive proximal weakness, markedly elevated CK, and myofiber necrosis with minimal inflammatory infiltrate on biopsy, triggered by autoantibodies against the HMGCR protein. 35 It occurs in roughly 2–3 per 100,000 statin users per year and, unlike ordinary SAMS, does not resolve with statin cessation; it requires immunosuppressive therapy, including intravenous immunoglobulin. 35
The proposed biochemical mechanism of statin myotoxicity is multifactorial: statins block mevalonate synthesis, depleting downstream isoprenoid intermediates (farnesyl and geranylgeranyl pyrophosphate) required for prenylation of small GTP-binding proteins such as Ras, Rho, and Rac, and they reduce synthesis of coenzyme Q10 (ubiquinone), which may impair mitochondrial respiration and increase reactive oxygen species. 30 These remain mechanistic hypotheses; notably, coenzyme Q10 supplementation has not reliably relieved statin myalgia in randomized trials, which should temper mechanistic overinterpretation. 30
Because systemic exposure is a primary determinant of myotoxic risk, statin choice, dose, and interactions are clinically relevant. Lipophilic statins metabolized by CYP3A4 (simvastatin, lovastatin, atorvastatin) are susceptible to interaction with strong CYP3A4 inhibitors such as amiodarone, diltiazem, macrolide antibiotics, azole antifungals, and cyclosporine; gemfibrozil should be avoided with all statins because it inhibits statin glucuronidation and OATP1B1-mediated hepatic uptake. 30, 31 Pravastatin, rosuvastatin, and the minimally CYP-metabolized pitavastatin are frequently used in patients with prior intolerance because of their dosing flexibility and favorable interaction profiles, although randomized evidence does not establish a universally lower SAMS rate based on hydrophilicity alone. 30
Host genetics also modulate this exposure. The transporter that carries statins into hepatocytes is encoded by SLCO1B1, and a common reduced-function variant (rs4149056, c.521T>C) increases systemic statin concentrations. In the SEARCH genome-wide study, the risk of myopathy in patients taking simvastatin 80 mg daily rose by an odds ratio of approximately 4.5 per copy of the C allele (with C-allele homozygotes at substantially higher absolute risk than heterozygotes). 57 The effect is strongest for simvastatin and attenuates for statins less dependent on OATP1B1 (such as rosuvastatin or pitavastatin); the 2022 Clinical Pharmacogenetics Implementation Consortium guideline translates SLCO1B1, ABCG2, and CYP2C9 genotype into actionable prescribing—favoring an alternative statin or a lower simvastatin dose in carriers. 57, 58 Pharmacogenomic testing is increasingly available and offers one route to distinguish patients at genuinely elevated pharmacologic risk from the much larger group whose reported symptoms are nocebo-mediated. 58
- Cleared by both hepatic and renal routes largely without cytochrome-P450 metabolism, which limits CYP-mediated drug–drug interactions. 31
- Despite high potency, its hydrophilic profile limits passive muscle penetration, and it undergoes only minimal CYP2C9 metabolism. 30, 31
- High bioavailability (≥ 60%) with minimal CYP metabolism (cleared mainly by biliary excretion) and a favorable muscle-tolerability and glucose profile. 30
Active individuals and endurance athletes report higher rates of SAMS, plausibly through the additive effect of exercise-induced muscle stress. A practical interpretive point deserves emphasis: creatine kinase (CK) elevations following prolonged or unaccustomed endurance exercise frequently exceed the values traditionally used to define statin myopathy, so a CK result in an athlete is best interpreted in light of recent exercise, repeat measurement after a rest interval, accompanying symptoms, and overall clinical context rather than a single threshold. Here the STOMP trial is reassuring: high-dose atorvastatin (80 mg) over six months in healthy, statin-naive subjects modestly increased muscle complaints and raised average CK by 20.8 U/L (P < 0.0001), but did not objectively impair muscle strength or exercise capacity. 36 For these patients, a low-dose hydrophilic statin (e.g., rosuvastatin 5 mg) or pitavastatin, with or without ezetimibe, is a rational strategy to preserve performance while lowering ApoB. 36
Statins are also associated with a small, dose-dependent increase in the risk of new-onset type 2 diabetes—on the order of ~0.1–0.2% per year of treatment, corresponding in the Sattar meta-analysis to roughly one additional case per 255 patients treated for four years—concentrated almost entirely in patients with pre-existing metabolic syndrome or pre-diabetes. 37 Pitavastatin is associated with a comparatively favorable glycemic profile and is a reasonable choice when this concern is prominent, though glycemic neutrality has not been conclusively established. 30
6. Non-Statin Pharmacotherapy: Targets, Efficacy, and Trials
When statin therapy alone is insufficient to meet lipid targets, or in cases of statin intolerance, non-statin therapies can further lower LDL-C and ApoB. 18 Their benefit, like that of statins, is proportional to the absolute reduction in atherogenic particles achieved, regardless of mechanism. 38
6.1 Ezetimibe
Ezetimibe binds the Niemann-Pick C1-Like 1 (NPC1L1) transporter on the brush-border membrane of enterocytes, inhibiting intestinal absorption of dietary and biliary cholesterol. 38 This reduces cholesterol delivery to the liver, depletes hepatic stores, and upregulates LDL receptors. Administered as an oral dose of 10 mg daily, ezetimibe reduces LDL-C by approximately 15–25% and ApoB by approximately 11–15%. In the landmark IMPROVE-IT trial, adding ezetimibe to simvastatin in patients after acute coronary syndrome reduced the primary endpoint from 34.7% to 32.7% (hazard ratio 0.936; 95% CI 0.89–0.99; P = 0.016)—a 6.4% relative reduction confirming that benefit is proportional to the absolute reduction in LDL-C. 38 Ezetimibe is well tolerated and is not associated with muscle toxicity or new-onset diabetes. 38
6.2 Bempedoic Acid
Bempedoic acid is an oral prodrug that inhibits ATP-citrate lyase (ACL), an enzyme upstream of HMG-CoA reductase in the cholesterol-biosynthesis pathway. 39 It requires activation by very-long-chain acyl-CoA synthetase-1 (ACSVL1), which is highly expressed in the liver but absent in skeletal muscle; this liver-specific activation minimizes muscle-related adverse effects, making bempedoic acid an option for statin-intolerant patients. 39 As monotherapy at 180 mg it reduces LDL-C by roughly 17–24% and ApoB by ~15%, and in fixed-dose combination with ezetimibe yields a synergistic reduction of roughly 38%. In the CLEAR Outcomes trial, which enrolled 13,970 statin-intolerant patients, bempedoic acid reduced the four-component MACE endpoint by 13% (hazard ratio 0.87; 95% CI 0.79–0.96; P = 0.004), with myocardial infarction reduced by 23% and coronary revascularization by 19%. 39 Its distinctive adverse effects are a rise in serum uric acid of roughly 0.8 mg/dL with a small excess of gout (about 3.1% versus 2.1% in CLEAR Outcomes) and a modest excess of tendon rupture (about 1.2% versus 0.9% in CLEAR Outcomes; a smaller absolute excess was seen in the earlier hypercholesterolemia trials); uric acid should be monitored periodically, particularly in patients with a history of gout. 39
6.3 Bile Acid Sequestrants
These agents bind bile acids in the intestine, preventing their reabsorption and forcing the liver to convert hepatic cholesterol into new bile acids, which upregulates LDL receptors. While effective at lowering LDL-C, their use is limited by gastrointestinal tolerability (constipation, flatulence) and by a tendency to raise triglycerides, so they are generally avoided in patients with significant hypertriglyceridemia. 51 Importantly, they can bind and reduce the absorption of other lipid-lowering drugs, so co-administered ezetimibe or bempedoic acid should be taken at least 2 hours before or 4 hours after a bile acid sequestrant. 39
6.4 PCSK9 Inhibitors: Monoclonal Antibodies Versus siRNA
PCSK9 is a hepatic serine protease that binds LDL receptors on the hepatocyte surface and directs them toward lysosomal degradation rather than recycling. 40 Inhibiting PCSK9 increases LDL-receptor density, enhancing clearance of LDL-C and ApoB. 40
- Evolocumab and Alirocumab. Fully human monoclonal antibodies that bind circulating PCSK9 in plasma, preventing its interaction with LDL receptors. Administered subcutaneously every 2 weeks or monthly, they reduce LDL-C by roughly 50–60% and ApoB by roughly 40–50%. Their cardiovascular benefit was demonstrated in the FOURIER (evolocumab) and ODYSSEY OUTCOMES (alirocumab) trials, each showing a ~15% reduction in major adverse cardiovascular events (hazard ratio 0.85); in ODYSSEY OUTCOMES, all-cause mortality was nominally lower with alirocumab (a prespecified but hierarchically secondary finding). Injection-site reactions (mild erythema, itching, or swelling) are the most common adverse event. 40, 41
- A small interfering RNA (siRNA) that inhibits synthesis of the PCSK9 protein intracellularly. Conjugated to triantennary N-acetylgalactosamine (GalNAc), it targets hepatic asialoglycoprotein receptors for selective hepatocyte uptake; the guide strand is then incorporated into the RNA-induced silencing complex (RISC), directing catalytic cleavage of PCSK9 messenger RNA. 42 Inclisiran is administered subcutaneously on Day 1, Day 90, and every 6 months thereafter—a twice-yearly maintenance schedule that can substantially improve long-term adherence. In the ORION-9, ORION-10, and ORION-11 trials it reduced LDL-C by roughly 48–52% and ApoB by roughly 38–45%, with a safety profile similar to placebo apart from mild, transient injection-site reactions. 42, 43 The event-driven ORION-4 outcomes trial remains ongoing, so its cardiovascular benefit awaits confirmation by hard endpoints. 42
6.5 Emerging Therapies
- A highly selective oral cholesteryl ester transfer protein (CETP) inhibitor. Earlier CETP inhibitors (torcetrapib, dalcetrapib, evacetrapib) failed in development because of off-target toxicity or inadequate efficacy; obicetrapib was specifically engineered to avoid those limitations, combining high selectivity with a favorable tolerability profile. It reduced LDL-C by approximately 30% in the BROADWAY trial of high-risk patients and by up to ~41% in the BROOKLYN heterozygous-FH trial, with ApoB reductions of roughly 20–24% and Lp(a) reductions of roughly 33–46%; the PREVAIL cardiovascular-outcomes trial is ongoing, so its effect on hard events is not yet established. 44, 45
- A ligand-conjugated antisense oligonucleotide that selectively inhibits apolipoprotein(a) synthesis in the liver, reducing circulating Lp(a) by up to ~80%; the Lp(a)HORIZON cardiovascular-outcomes trial is underway. 46
- Olpasiran and Lepodisiran. Hepatocyte-directed siRNAs that silence the LPA gene, reducing hepatic Lp(a) production by up to ~94–97%; both are in phase 3 outcomes evaluation (OCEAN[a]-Outcomes and ACCLAIM-Lp[a]). 47, 48
6.6 Residual Inflammatory Risk: A Complementary Target
Even with ApoB driven to low levels, a substantial residual risk of events persists, and a major component of it is inflammatory—consistent with the biology in which the inflammatory response is largely downstream of, but not identical to, lipoprotein retention. 52 Three landmark randomized trials established inflammation as a modifiable target independent of lipid lowering. In CANTOS, the interleukin-1β antibody canakinumab reduced major adverse cardiovascular events in patients with prior myocardial infarction and high-sensitivity C-reactive protein ≥ 2 mg/L, without lowering LDL-C—the first proof that targeting inflammation alone reduces events. 59 Because canakinumab is costly and raised fatal infections, attention shifted to low-dose colchicine: COLCOT (after recent myocardial infarction) and LoDoCo2 (in chronic coronary disease) each reduced events by roughly 23–31% on top of statin therapy, and a low-dose colchicine formulation was subsequently approved for cardiovascular risk reduction. 60, 61 These agents do not lower ApoB and are not substitutes for it; they define a complementary axis of treatment for patients who remain at high risk despite well-controlled lipoproteins. Their efficacy is fully compatible with—though it does not by itself prove—the model in which retained ApoB particles initiate the inflammatory cascade that these therapies interrupt downstream. 52, 59 Attention is now turning further upstream to interleukin-6 (IL-6) signaling, a central node in the residual inflammatory pathway, which is also being investigated as a potential therapeutic target. 52
7. Specific Clinical Management of Special Populations
- Familial Hypercholesterolemia (FH). HeFH requires early initiation of high-intensity statins, often with ezetimibe and PCSK9-targeted therapy, to achieve intensive LDL-C targets. 18, 20 In HoFH, extreme LDL-C elevation drives accelerated atherogenesis, and LDL-receptor deficiency blunts standard therapy. 21 Evinacumab, an angiopoietin-like 3 (ANGPTL3) inhibitor that lowers LDL-C by roughly 47% through an LDL-receptor-independent pathway, reduced LDL-C by that margin in the ELIPSE HoFH trial and is approved to progressively younger pediatric ages; lomitapide, a microsomal triglyceride transfer protein (MTP) inhibitor, lowers LDL-C by roughly 50%. 49, 50
- Statin Intolerance. True, rechallenge-confirmed statin intolerance affects only about 5–10% of patients. 32 For these individuals, combination non-statin therapy is highly effective: a regimen of ezetimibe, bempedoic acid, and inclisiran can lower LDL-C by well over 50% and has been used successfully even in patients with a history of statin-induced rhabdomyolysis. 39, 42
- Endurance Athletes. Highly active individuals may report SAMS or myalgia, particularly on high-dose lipophilic statins. A clinically important distinction is often blurred here: strenuous exercise, especially eccentric or unaccustomed exertion, transiently raises creatine kinase (CK) into ranges that can reach many multiples of the upper limit of normal in healthy athletes, and this exercise-induced elevation must not be mistaken for statin-associated muscle injury. 31 True statin myopathy is distinguished by persistent proximal weakness or symptoms out of proportion to training load and by CK elevation that does not normalize with rest; a CK drawn shortly after hard training, by contrast, is expected to be high and typically resolves within days, so repeating CK after a rest interval and correlating with symptoms avoids unnecessary statin discontinuation. 31 Low-dose rosuvastatin (5 mg), pravastatin, or pitavastatin are preferred to preserve performance while lowering ApoB, and the STOMP trial found no objective decrement in muscle strength or exercise capacity even with high-dose atorvastatin over six months. 36
- Women and Pregnancy. Reproductive risk markers—early menopause (< 45 years), preeclampsia, gestational diabetes, or gestational hypertension—should be considered when personalizing risk. Statins are usually avoided before conception and during pregnancy and lactation; however, the U.S. Food and Drug Administration removed the blanket contraindication in 2021, and selected patients at very high cardiovascular risk—for example, those with homozygous familial hypercholesterolemia or established ASCVD—may warrant individualized specialist management rather than automatic discontinuation. 18
- Diabetes and Inflammatory Diseases. Patients with type 2 diabetes or chronic inflammatory conditions (rheumatoid arthritis, psoriasis) have accelerated vascular aging and higher baseline risk. ApoB should be targeted intensively given the atherogenicity of small, dense LDL and VLDL, with residual inflammatory risk addressed alongside it. 8, 52
- Very High Coronary Artery Calcium (CAC). A high CAC score reclassifies a patient into a very-high-risk category warranting an intensive LDL-C target, whereas a CAC of 0 in the absence of other high-risk features (diabetes, smoking, severe hypercholesterolemia) may permit deferral of therapy with reassessment in several years. 18
8. Clinical Decision Framework and Algorithmic Pathway
The sequence below is illustrative rather than mandatory. Selection and order of therapy should reflect baseline risk, the magnitude and urgency of the required LDL-C or ApoB reduction, established ASCVD or familial hypercholesterolemia, statin tolerance, cost, access, and patient preference—for example, a patient far from goal or at very high risk may warrant earlier escalation to PCSK9-targeted therapy rather than strict stepwise progression. With that caveat, the following stepwise framework organizes the available options.
- Lifestyle Optimization. Initiate a Mediterranean-style or plant-based pattern, saturated fat < 7% of daily energy, viscous fiber, and plant sterols (2 g/day), with aerobic (≥ 150 min/week) and resistance exercise. Appropriate alone for low-risk primary prevention, but pharmacotherapy should not be delayed in patients with high baseline risk, genetic hypercholesterolemia, or established subclinical plaque. 18, 28
- First-Line Statin. Rosuvastatin or atorvastatin for standard high-intensity lowering (targeting ≥ 50% LDL-C reduction); pravastatin or low-dose rosuvastatin for athletes and frail older adults; pitavastatin when a comparatively favorable glycemic profile is a priority. 30, 36
- Secondary Oral Intensification. If LDL-C or ApoB remains above target on maximally tolerated statin, add ezetimibe 10 mg—effective, well tolerated, and cost-effective. 38
- Statin-Sparing Optimization. For persistent SAMS or additional oral lowering, add bempedoic acid, which avoids skeletal-muscle activation; monitor uric acid, particularly in patients with a history of gout. 39
- Advanced Biologic Therapy. For clinical ASCVD, HeFH, or extreme hypercholesterolemia not at goal on oral therapy, add a PCSK9 monoclonal antibody (for rapid, maximal lowering with self-injection) or inclisiran (for twice-yearly, adherence-friendly dosing). 40, 42
9. Quantitative Summary Tables
Table 1. Pharmacokinetic and Pharmacodynamic Profiles of Major Statins
| Parameter | Atorva | Rosuva | Prava | Pitava | Simva | Lova | Fluva |
| Solubility | Lipophilic | Hydrophilic | Hydrophilic | Lipophilic | Lipophilic | Lipophilic | Lipophilic |
| Bioavailability | ~12% | ~20% | ~18% | ≥60% | <5% | ~5% | ~24% |
| Clearance | CYP3A4 | CYP2C9 (min.) | Non-CYP renal | Minimal CYP | CYP3A4 | CYP3A4 | CYP2C9 |
| Half-life (h) | 14 | 19 | 1.8 | 11 | 2–5 | 2–5 | 1.2 |
| Major interaction route | CYP3A4/OATP1B1 | OATP1B1 (min. CYP) | None (renal) | OATP1B1 (min. CYP) | CYP3A4/OATP1B1 | CYP3A4/OATP1B1 | CYP2C9/OATP1B1 |
| SLCO1B1 sensitivity* | Moderate | Low | Low | Low | High | Moderate | Low |
| LDL-C reduction | 37–57% | 45–63% | 20–35% | 30–45% | 20–45% | 20–40% | 20–35% |
| ApoB reduction | 30–45% | 35–50% | 15–25% | 25–35% | 15–35% | 15–30% | 15–25% |
Sources and definitions. Solubility, bioavailability, hepatic clearance pathway, and plasma half-life are drawn from primary pharmacokinetic characterization [30] and standard reference summaries [31]. LDL-C and ApoB reduction ranges reflect dose-dependent effects reported in trials and product labeling; ApoB reductions are typically a few percentage points smaller than the corresponding LDL-C reduction.
*SLCO1B1 sensitivity denotes the extent to which reduced-function OATP1B1 variants raise systemic exposure and myopathy risk for that agent, as codified in the 2022 CPIC guideline; it is highest for simvastatin. This table reports pharmacokinetic and pharmacogenetic properties rather than a validated comparative clinical ranking of muscle-symptom or diabetes risk, both of which depend more on dose, systemic exposure, drug interactions, and individual susceptibility than on drug identity alone (see Section 5.1). [31], [58]
Table 2. Comparative Efficacy and Safety of Non-Statin Therapies
| Parameter | Ezetimibe | Bempedoic acid | Evolocumab / Alirocumab | Inclisiran |
| Mechanism | NPC1L1 inhibition | ATP-citrate lyase inhibition | Circulating PCSK9 neutralization | PCSK9 mRNA silencing (siRNA) |
| LDL-C reduction | 15–25% | 17–24% (~38% + ezet.) | 50–60% | ~50% |
| ApoB reduction | 11–15% | ~15% (25–30% + ezet.) | 40–50% | 38–45% |
| Relative ↓ in trial’s 1° CV endpoint | 6.4% (IMPROVE-IT) | 13% (CLEAR Outcomes) | ~15% (FOURIER / ODYSSEY) | Ongoing (ORION-4) |
| Absolute risk reduction / follow-up | ~2.0% / ~7 yr | ~1.6% / 3.4 yr | ~1.5% / 2.2 yr | Not yet established |
| Muscle symptoms | Neutral | Similar to placebo / low excess | Neutral | Neutral |
| Other notable adverse effects | Neutral | Hyperuricemia, gout, cholelithiasis; uncommon tendon-rupture warning | Injection-site reactions | Injection-site reactions |
| Glycemia | Neutral | Neutral | Neutral | Neutral |
| Uric acid / gout | Neutral | ↑ ~0.8 mg/dL; gout ~3% vs 2% (CLEAR) | Neutral | Neutral |
| Route / frequency | Oral; daily | Oral; daily | SC; every 2–4 weeks | SC; day 1, 90, then q6 mo |
| Cost | Low (generic) | Moderate | High | High |
SC = subcutaneous. Relative and absolute effects are drawn from the pivotal trials: ezetimibe from IMPROVE-IT (post-ACS; ~7-year follow-up; secondary prevention) [38]; bempedoic acid from CLEAR Outcomes (statin-intolerant; median 3.4-year follow-up; primary 4-component endpoint) [39]; evolocumab/alirocumab from FOURIER and ODYSSEY OUTCOMES (established ASCVD/post-ACS; ~2.2–2.8-year follow-up) [40], [41]; inclisiran lipid effects from ORION-9/-10/-11, with the cardiovascular-outcome endpoint under evaluation in ORION-4 [42], [43]. The tendon-rupture warning derives from the CLEAR Outcomes safety data.
Because the trials differ in population, baseline risk, follow-up duration, and background therapy, the relative MACE reductions and NNT estimates are not directly comparable across columns; they should be read within the context of each trial rather than as head-to-head equivalents.
Table 3. Comparison of Major Lipid-Management Guidelines
| Domain | 2026 ACC/AHA/Multisociety | ESC/EAS 2019 | CCS 2021 | Pediatric (NHLBI/EAS) |
| Risk tool | PREVENT equations (10- & 30-yr) | SCORE2 / SCORE2-OP | Modified FRS | Family history + LDL-C level |
| Preferred marker | LDL-C; ApoB & Lp(a) emphasized | LDL-C primary; ApoB, non-HDL-C | Non-HDL-C or ApoB if TG > 1.5 mmol/L | LDL-C |
| 2° prevention LDL-C goal | < 55 mg/dL (very high risk) | < 55 mg/dL (& ≥ 50% ↓) | ≤ 1.8 mmol/L (~70 mg/dL) or ≥ 50% ↓ | n/a |
| Universal Lp(a) | Yes, once in life | Once in life | Once in life | Consider with family history |
| Screening start | Youth; young adults if LDL-C ≥ 160 | Men ≥ 40, women ≥ 50 / post-menopausal | All adults ≥ 40 (earlier if risk) | Universal 9–11 & 17–21 y |
| FH statin start | Youth with FH (early) | From age 8–10 | On diagnosis | Age 8–10 (≥50% ↓); target < 3.5 mmol/L from 10 y |
| Distinctive feature | Restores absolute goals; lifespan/early emphasis | Lower-is-better; risk-based tiers | Health-behaviour + statin-indicated conditions | Pubertal timing; specialist-led |
FRS = Framingham Risk Score; FH = familial hypercholesterolemia; TG = triglycerides; ↓ = reduction. Entries summarize headline positions and are not a substitute for the full guideline text. Sources: 2026 ACC/AHA/Multisociety [18]; ESC/EAS 2019 [51]; CCS 2021 [24]; pediatric NHLBI [19] and EAS [20]. Some ESC/EAS and CCS figures reflect the most recent published versions and may be updated by subsequent focused updates.
10. Conclusions: A Reasoned Case for Earlier, Lower, and Longer
The evidence assembled here supports a coherent proposition, offered here with its inferential limits made explicit. Because ApoB-containing lipoproteins are, under the response-to-retention framework, the necessary initiating agents of atherosclerosis, because their retention within the arterial wall sets off the inflammatory cascade that becomes plaque, and because the resulting disease remains the leading cause of death worldwide, the totality of current evidence suggests that many patients may benefit from earlier, lower, and more sustained ApoB reduction than is commonly achieved in contemporary practice. 1, 3, 12 The cumulative-exposure framework reframes the clinical question from “is this person high-risk today?” to “how many ApoB-years will this person accumulate over a lifetime?”—and the genetic evidence indicates that the answer to the second question, addressed early, is associated with protection that late intervention has not been shown to match. 13, 14, 17 It is important to be candid that this case rests substantially on mechanistic, genetic, and observational evidence together with extrapolation from shorter-term trials; it is not established by decades-long randomized trials of early treatment in low-risk young adults, which do not exist.
This is not a call to medicate indiscriminately, and the argument is reasonable precisely because it survives an honest accounting of harm. In patients who report intolerance, the muscle symptoms that most deter treatment are largely reproduced by placebo; the objectively defined myopathies are rare; rhabdomyolysis occurs only a few times per hundred thousand person-years; and the new-onset-diabetes signal is small and concentrated in those already dysglycemic. 32, 34, 37 Against these modest and largely manageable harms sit absolute event reductions that scale with baseline risk and with duration of exposure. The rule that emerges is one of calibrated intensity rather than uniform aggressiveness: where estimated risk is low, benefit and harm are close and the decision is genuinely preference-sensitive, belonging to the informed patient; as risk, genetic burden, or documented subclinical plaque rises—and particularly when exposure begins early in life—the expected benefit increasingly exceeds the expected harm. 16, 18, 37 For the individual with familial hypercholesterolemia, a strong family history, elevated Lp(a), or plaque on imaging, the evidence provides a strong rationale not to defer treatment, weighed through shared decision-making.
Modern therapeutics make this achievable with an acceptable safety profile. Alternative statin selection, lower or intermittent dosing, and combination therapy can often permit clinically meaningful lipid lowering in patients with prior muscle symptoms; ezetimibe and bempedoic acid deliver substantial ApoB lowering without meaningful muscle risk; and twice-yearly inclisiran offers durable control for patients who struggle with adherence. 30, 38, 39, 42 Several clinical areas nonetheless require further research: confirming the long-term cardiovascular outcomes of siRNA-based and Lp(a)-directed therapies in event-driven trials (for example, ORION-4 and the ongoing Lp(a) outcomes trials); evaluating the net benefit, safety, cost-effectiveness, and acceptability of initiating therapy earlier in lower-risk and pediatric cohorts; and standardizing direct ApoB and Lp(a) measurement globally so that the patients who would benefit most from early intervention can be identified before their arteries have kept the record for them. 13, 46
11. Limitations
Several limitations should temper interpretation of this review and of the recommendations drawn from it.
- Evidence hierarchy and inference. Much of the argument for earlier intervention across the life course rests on mechanistic evidence, Mendelian randomization, observational cohorts, and extrapolation from shorter-term randomized trials rather than on randomized trials that initiate therapy in low-risk young adults and follow them for decades. Mendelian randomization estimates the effect of lifelong genetically-proxied exposure, which is not numerically interchangeable with the effect of a drug started in midlife; treating the two as equivalent would overstate achievable benefit. 14, 17
- Generalizability and thresholds. Treatment thresholds and benefit-to-harm ratios are population- and context-dependent. The cumulative-exposure model and the illustrative “ApoB-years” trajectories are heuristics that organize the evidence; they are not validated individual-level risk calculators, and the specific age projections are model-derived rather than directly measured. 13
- Cost, access, adherence, and equity. The real-world value of earlier and longer lipid lowering depends not only on biologic efficacy but on affordability, access, lifelong adherence, and the potential burdens of medicalization—factors that fall unevenly across populations and that this review does not quantify. The expanding availability of generic statins and ezetimibe has markedly improved affordability, whereas PCSK9 inhibitors, inclisiran, and emerging Lp(a)-directed therapies remain substantially more expensive and may be constrained by insurance coverage or health-system resources; cost-effectiveness therefore depends strongly on baseline cardiovascular risk, and cost-related nonadherence remains a measurable barrier to sustained benefit. 56
- Incomplete outcomes data for newer agents. For several therapies discussed—inclisiran, obicetrapib, and the Lp(a)-directed siRNA and antisense agents—robust lipid-lowering and biomarker data are available, but confirmatory hard-outcome trials are ongoing; their cardiovascular benefit should be regarded as expected rather than established. 42, 44, 46
- Special populations. Evidence in pregnancy, lactation, very young pediatric cohorts, and the frail elderly remains comparatively limited, and recommendations in these groups rely more heavily on expert consensus and individualized specialist judgment than on large randomized trials. 19, 21
- Methodological scope. As a narrative review, this article did not apply a formal search protocol or risk-of-bias appraisal, and its selection and emphasis of evidence necessarily reflect editorial judgment; readers should interpret its conclusions accordingly and consult primary guidelines for individual clinical decisions. 53, 54
References
- Mensah GA, Fuster V, Murray CJL, Roth GA; Global Burden of Cardiovascular Diseases and Risks Collaborators. Global Burden of Cardiovascular Diseases and Risks, 1990-2022. J Am Coll Cardiol. 2023;82(25):2350-2473. doi:10.1016/j.jacc.2023.11.007
- World Health Organization, “Cardiovascular diseases (CVDs),” WHO Fact Sheet, updated Jul. 31, 2025. [Online]. Available: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed Jul. 12, 2026).
- Williams KJ, Tabas I. The response-to-retention hypothesis of early atherogenesis. Arterioscler Thromb Vasc Biol. 1995;15(5):551-561. doi:10.1161/01.atv.15.5.551
- Tabas I, Williams KJ, Borén J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 2007;116(16):1832-1844. doi:10.1161/CIRCULATIONAHA.106.676890
- Kraehling JR, Chidlow JH, Rajagopal C, et al. Genome-wide RNAi screen reveals ALK1 mediates LDL uptake and transcytosis in endothelial cells. Nat Commun. 2016;7:13516. Published 2016 Nov 21. doi:10.1038/ncomms13516
- Huang L, Chambliss KL, Gao X, et al. SR-B1 drives endothelial cell LDL transcytosis via DOCK4 to promote atherosclerosis. Nature. 2019;569(7757):565-569. doi:10.1038/s41586-019-1140-4
- Tabas I, Li Y, Brocia RW, Xu SW, Swenson TL, Williams KJ. Lipoprotein lipase and sphingomyelinase synergistically enhance the association of atherogenic lipoproteins with smooth muscle cells and extracellular matrix. A possible mechanism for low density lipoprotein and lipoprotein(a) retention and macrophage foam cell formation. J Biol Chem. 1993;268(27):20419-20432.
- Sniderman AD, Thanassoulis G, Glavinovic T, et al. Apolipoprotein B Particles and Cardiovascular Disease: A Narrative Review. JAMA Cardiol. 2019;4(12):1287-1295. doi:10.1001/jamacardio.2019.3780
- Ference BA, Kastelein JJP, Ray KK, et al. Association of Triglyceride-Lowering LPL Variants and LDL-C-Lowering LDLR Variants With Risk of Coronary Heart Disease. JAMA. 2019;321(4):364-373. doi:10.1001/jama.2018.20045
- Richardson TG, Sanderson E, Palmer TM, et al. Evaluating the relationship between circulating lipoprotein lipids and apolipoproteins with risk of coronary heart disease: A multivariable Mendelian randomisation analysis. PLoS Med. 2020;17(3):e1003062. Published 2020 Mar 23. doi:10.1371/journal.pmed.1003062
- Zuber V, Gill D, Ala-Korpela M, et al. High-throughput multivariable Mendelian randomization analysis prioritizes apolipoprotein B as key lipid risk factor for coronary artery disease. Int J Epidemiol. 2021;50(3):893-901. doi:10.1093/ije/dyaa216
- Ference BA, Ginsberg HN, Graham I, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2017;38(32):2459-2472. doi:10.1093/eurheartj/ehx144
- Ference BA, Braunwald E, Catapano AL. The LDL cumulative exposure hypothesis: evidence and practical applications. Nat Rev Cardiol. 2024;21(10):701-716. doi:10.1038/s41569-024-01039-5
- Ference BA, Yoo W, Alesh I, et al. Effect of long-term exposure to lower low-density lipoprotein cholesterol beginning early in life on the risk of coronary heart disease: a Mendelian randomization analysis. J Am Coll Cardiol. 2012;60(25):2631-2639. doi:10.1016/j.jacc.2012.09.017
- Cholesterol Treatment Trialists’ (CTT) Collaboration, Baigent C, Blackwell L, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376(9753):1670-1681. doi:10.1016/S0140-6736(10)61350-5
- Cholesterol Treatment Trialists’ (CTT) Collaborators, Mihaylova B, Emberson J, et al. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta-analysis of individual data from 27 randomised trials. Lancet. 2012;380(9841):581-590. doi:10.1016/S0140-6736(12)60367-5
- Daghlas I, Gill D. Low-density lipoprotein cholesterol and lifespan: A Mendelian randomization study. Br J Clin Pharmacol. 2021;87(10):3916-3924. doi:10.1111/bcp.14811
- Writing Committee Members, Blumenthal RS, Morris PB, et al. 2026 ACC/AHA/AACVPR/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Dyslipidemia: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2026;153(17):e1154-e1276. doi:10.1161/CIR.0000000000001423
- Expert Panel on Integrated Guidelines for Cardiovascular Health and Risk Reduction in Children and Adolescents; National Heart, Lung, and Blood Institute. Expert panel on integrated guidelines for cardiovascular health and risk reduction in children and adolescents: summary report. Pediatrics. 2011;128 Suppl 5(Suppl 5):S213-S256. doi:10.1542/peds.2009-2107C
- Wiegman A, Gidding SS, Watts GF, et al. Familial hypercholesterolaemia in children and adolescents: gaining decades of life by optimizing detection and treatment. Eur Heart J. 2015;36(36):2425-2437. doi:10.1093/eurheartj/ehv157
- Cuchel M, Bruckert E, Ginsberg HN, et al. Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur Heart J. 2014;35(32):2146-2157. doi:10.1093/eurheartj/ehu274
- Pletcher MJ, Bibbins-Domingo K, Liu K, et al. Nonoptimal lipids commonly present in young adults and coronary calcium later in life: the CARDIA (Coronary Artery Risk Development in Young Adults) study. Ann Intern Med. 2010;153(3):137-146. doi:10.7326/0003-4819-153-3-201008030-00004
- Zheutlin AR, Handoo F, Luebbe S, et al. Cumulative exposure to atherogenic lipoprotein particles in young adults and subsequent incident atherosclerotic cardiovascular disease. Eur Heart J. 2025;46(41):4302-4312. doi:10.1093/eurheartj/ehaf472
- Pearson GJ, Thanassoulis G, Anderson TJ, et al. 2021 Canadian Cardiovascular Society Guidelines for the Management of Dyslipidemia for the Prevention of Cardiovascular Disease in Adults. Can J Cardiol. 2021;37(8):1129-1150. doi:10.1016/j.cjca.2021.03.016
- Chiavaroli L, Nishi SK, Khan TA, et al. Portfolio Dietary Pattern and Cardiovascular Disease: A Systematic Review and Meta-analysis of Controlled Trials. Prog Cardiovasc Dis. 2018;61(1):43-53. doi:10.1016/j.pcad.2018.05.004
- Brown L, Rosner B, Willett WW, Sacks FM. Cholesterol-lowering effects of dietary fiber: a meta-analysis. Am J Clin Nutr. 1999;69(1):30-42. doi:10.1093/ajcn/69.1.30
- Ras RT, Geleijnse JM, Trautwein EA. LDL-cholesterol-lowering effect of plant sterols and stanols across different dose ranges: a meta-analysis of randomised controlled studies. Br J Nutr. 2014;112(2):214-219. doi:10.1017/S0007114514000750
- Sacks FM, Lichtenstein AH, Wu JHY, et al. Dietary Fats and Cardiovascular Disease: A Presidential Advisory From the American Heart Association. Circulation. 2017;136(3):e1-e23. doi:10.1161/CIR.0000000000000510
- Bull FC, Al-Ansari SS, Biddle S, et al. World Health Organization 2020 guidelines on physical activity and sedentary behaviour. Br J Sports Med. 2020;54(24):1451-1462. doi:10.1136/bjsports-2020-102955
- Schachter M. Chemical, pharmacokinetic and pharmacodynamic properties of statins: an update. Fundam Clin Pharmacol. 2005;19(1):117-125. doi:10.1111/j.1472-8206.2004.00299.x
- Thompson PD, Panza G, Zaleski A, Taylor B. Statin-Associated Side Effects. J Am Coll Cardiol. 2016;67(20):2395-2410. doi:10.1016/j.jacc.2016.02.071
- Howard JP, Wood FA, Finegold JA, et al. Side Effect Patterns in a Crossover Trial of Statin, Placebo, and No Treatment. J Am Coll Cardiol. 2021;78(12):1210-1222. doi:10.1016/j.jacc.2021.07.022
- Wood FA, Howard JP, Finegold JA, et al. N-of-1 Trial of a Statin, Placebo, or No Treatment to Assess Side Effects. N Engl J Med. 2020;383(22):2182-2184. doi:10.1056/NEJMc2031173
- Graham DJ, Staffa JA, Shatin D, et al. Incidence of hospitalized rhabdomyolysis in patients treated with lipid-lowering drugs. JAMA. 2004;292(21):2585-2590. doi:10.1001/jama.292.21.2585
- Khoo T, Tan E, Limaye V, et al. The incidence of anti-HMGCR immune-mediated necrotizing myopathy: an Australian and UK retrospective multi-site cohort study. Rheumatology (Oxford). 2025;64(9):4995-5003. doi:10.1093/rheumatology/keaf238
- Parker BA, Capizzi JA, Grimaldi AS, et al. Effect of statins on skeletal muscle function. Circulation. 2013;127(1):96-103. doi:10.1161/CIRCULATIONAHA.112.136101
- Sattar N, Preiss D, Murray HM, et al. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet. 2010;375(9716):735-742. doi:10.1016/S0140-6736(09)61965-6
- Cannon CP, Blazing MA, Giugliano RP, et al. Ezetimibe Added to Statin Therapy after Acute Coronary Syndromes. N Engl J Med. 2015;372(25):2387-2397. doi:10.1056/NEJMoa1410489
- Nissen SE, Lincoff AM, Brennan D, et al. Bempedoic Acid and Cardiovascular Outcomes in Statin-Intolerant Patients. N Engl J Med. 2023;388(15):1353-1364. doi:10.1056/NEJMoa2215024
- Sabatine MS, Giugliano RP, Keech AC, et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N Engl J Med. 2017;376(18):1713-1722. doi:10.1056/NEJMoa1615664
- Schwartz GG, Steg PG, Szarek M, et al. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N Engl J Med. 2018;379(22):2097-2107. doi:10.1056/NEJMoa1801174
- Ray KK, Wright RS, Kallend D, et al. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N Engl J Med. 2020;382(16):1507-1519. doi:10.1056/NEJMoa1912387
- Raal FJ, Kallend D, Ray KK, et al. Inclisiran for the Treatment of Heterozygous Familial Hypercholesterolemia. N Engl J Med. 2020;382(16):1520-1530. doi:10.1056/NEJMoa1913805
- Nicholls SJ, Nelson AJ, Ditmarsch M, et al. Safety and Efficacy of Obicetrapib in Patients at High Cardiovascular Risk. N Engl J Med. 2025;393(1):51-61. doi:10.1056/NEJMoa2415820
- Nicholls SJ, Nelson AJ, Ditmarsch M, et al. Obicetrapib in patients with heterozygous familial hypercholesterolemia: the BROOKLYN randomized clinical trial. Nat Med. 2026;32(3):1052-1060. doi:10.1038/s41591-025-04179-4
- Tsimikas S, Karwatowska-Prokopczuk E, Gouni-Berthold I, et al. Lipoprotein(a) Reduction in Persons with Cardiovascular Disease. N Engl J Med. 2020;382(3):244-255. doi:10.1056/NEJMoa1905239
- O’Donoghue ML, Rosenson RS, Gencer B, et al. Small Interfering RNA to Reduce Lipoprotein(a) in Cardiovascular Disease. N Engl J Med. 2022;387(20):1855-1864. doi:10.1056/NEJMoa2211023
- Nissen SE, Ni W, Shen X, et al. Lepodisiran – A Long-Duration Small Interfering RNA Targeting Lipoprotein(a). N Engl J Med. 2025;392(17):1673-1683. doi:10.1056/NEJMoa2415818
- Raal FJ, Rosenson RS, Reeskamp LF, et al. Evinacumab for Homozygous Familial Hypercholesterolemia. N Engl J Med. 2020;383(8):711-720. doi:10.1056/NEJMoa2004215
- Cuchel M, Meagher EA, du Toit Theron H, et al. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: a single-arm, open-label, phase 3 study. Lancet. 2013;381(9860):40-46. doi:10.1016/S0140-6736(12)61731-0
- Mach F, Baigent C, Catapano AL, et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. 2020;41(1):111-188. doi:10.1093/eurheartj/ehz455
- Ridker PM, Bhatt DL, Pradhan AD, et al. Inflammation and cholesterol as predictors of cardiovascular events among patients receiving statin therapy: a collaborative analysis of three randomised trials. Lancet. 2023;401(10384):1293-1301. doi:10.1016/S0140-6736(23)00215-5
- Page MJ, McKenzie JE, Bossuyt PM, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 2021;372:n71. Published 2021 Mar 29. doi:10.1136/bmj.n71
- Shea BJ, Reeves BC, Wells G, et al. AMSTAR 2: a critical appraisal tool for systematic reviews that include randomised or non-randomised studies of healthcare interventions, or both. BMJ. 2017;358:j4008. Published 2017 Sep 21. doi:10.1136/bmj.j4008
- Borén J, Chapman MJ, Krauss RM, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2020;41(24):2313-2330. doi:10.1093/eurheartj/ehz962
- Khera R, Valero-Elizondo J, Das SR, et al. Cost-Related Medication Nonadherence in Adults With Atherosclerotic Cardiovascular Disease in the United States, 2013 to 2017. Circulation. 2019;140(25):2067-2075. doi:10.1161/CIRCULATIONAHA.119.041974
- SEARCH Collaborative Group, Link E, Parish S, et al. SLCO1B1 variants and statin-induced myopathy–a genomewide study. N Engl J Med. 2008;359(8):789-799. doi:10.1056/NEJMoa0801936
- Cooper-DeHoff RM, Niemi M, Ramsey LB, et al. The Clinical Pharmacogenetics Implementation Consortium Guideline for SLCO1B1, ABCG2, and CYP2C9 genotypes and Statin-Associated Musculoskeletal Symptoms. Clin Pharmacol Ther. 2022;111(5):1007-1021. doi:10.1002/cpt.2557
- Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med. 2017;377(12):1119-1131. doi:10.1056/NEJMoa1707914
- Tardif JC, Kouz S, Waters DD, et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N Engl J Med. 2019;381(26):2497-2505. doi:10.1056/NEJMoa1912388
- Nidorf SM, Fiolet ATL, Mosterd A, et al. Colchicine in Patients with Chronic Coronary Disease. N Engl J Med. 2020;383(19):1838-1847. doi:10.1056/NEJMoa2021372
- Nordmann AJ, Suter-Zimmermann K, Bucher HC, et al. Meta-analysis comparing Mediterranean to low-fat diets for modification of cardiovascular risk factors. Am J Med. 2011;124(9):841-51.e2. doi:10.1016/j.amjmed.2011.04.024
- Koch CA, Kjeldsen EW, Frikke-Schmidt R. Vegetarian or vegan diets and blood lipids: a meta-analysis of randomized trials. Eur Heart J. 2023;44(28):2609-2622. doi:10.1093/eurheartj/ehad211