
Picture the scene: a quiet Saturday morning, the kitchen filled with the heavy, savory scent of searing ribeye and the rhythmic sizzle of butter-poached eggs. For decades, this “steak and eggs” breakfast has been cast as the ultimate dietary villain—a shortcut to a heart attack. Recently, scientists thought they had finally caught the culprit red-handed. They found a molecule in our blood called Trimethylamine N-Oxide, or TMAO.
The early data looked damning. When TMAO levels went up, heart health seemed to crater. It looked like a “smoking gun.” But as we dig into a massive new review of the evidence, the story takes a turn into the strange. It turns out that focusing on TMAO as a “killer” might be like blaming a smoke detector for a house fire. The detector makes a lot of noise when things go wrong, but it isn’t the thing that’s burning.
Is TMAO a direct metabolic assassin, or is it just the world’s most sensitive reporter for a body in trouble? From the “Fish Paradox” to the “Broken Filter” problem, let’s look at the hidden truths flipping the script on heart science.
1. Your Gut is a Chemical Factory (and Your Liver is the Refiner)
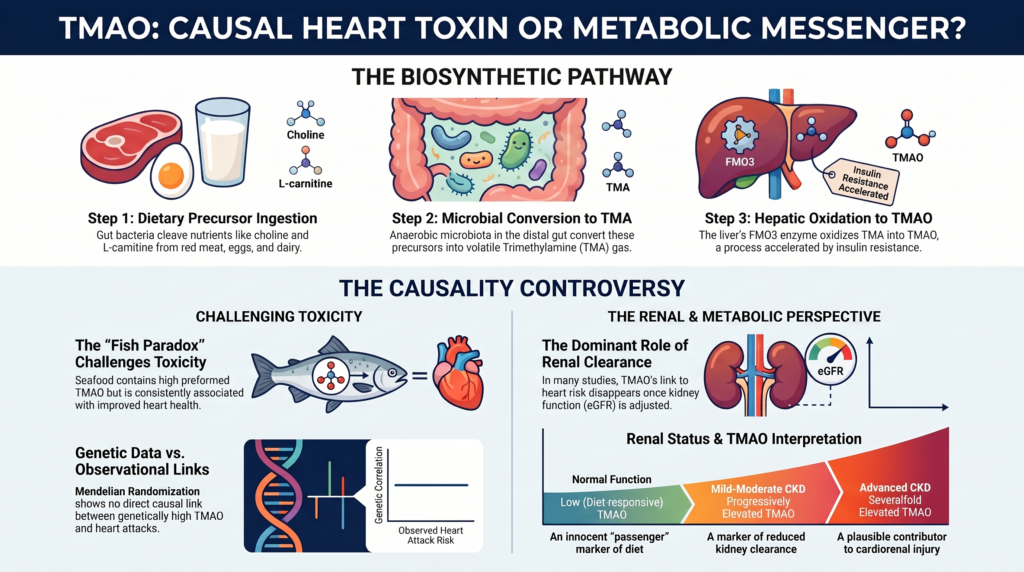
TMAO doesn’t just float into your bloodstream by magic. It is the end product of a high-tech, multi-step manufacturing process that involves you, your “tenants” (gut bacteria), and your liver. You can think of it as a three-stage industrial plant:
- The Raw Materials: You eat foods rich in nutrients like choline and L-carnitine. These are found in red meat, egg yolks, and full-fat dairy.
- The Gut Assembly Line: This is where things get interesting. Some of these nutrients escape your own digestion and reach the “factory floor” of your gut, where trillions of bacteria live. These bacteria have a specific job: they “cleave”—or chop—the chemical bonds of your food to create a stinky, volatile gas called TMA (trimethylamine).
- The Liver Refinery: This gas is lipophilic, meaning it easily slips through your gut wall and hitches a ride to the liver. There, a “refining machine” (an enzyme called FMO3) turns that smelly gas into TMAO—which is odorless and water-soluble, making it easy for the body to move around.
“Anaerobic microbiota cleave the carbon–nitrogen bonds of the precursors, releasing the volatile gas trimethylamine (TMA).” — Source Evidence Review
This “meta-organismal” process means your TMAO levels aren’t just a tally of your steak intake; they are a reflection of how many “factory workers” you have in your gut and how hard your liver refinery is working.
2. The “Fish Paradox” That Flips the Script
If TMAO were a direct poison that causes “rust” in our arteries, then eating foods that spike TMAO should be a death sentence. This is where the story hits a massive snag known as the “Fish Paradox.”
In the wild, marine fish like cod and halibut use pure, pre-formed TMAO to protect their cells from the crushing pressure and salt of the deep ocean. When you eat a piece of fish, you aren’t waiting for your gut bacteria to build TMAO from scratch. You are absorbing it directly into your blood.
The difference in scale is mind-blowing. Eating a steak might give you a modest bump in TMAO. But a single meal of cod or halibut can spike your TMAO levels thousands of µmol/L into the stratosphere. For comparison, meat-eaters typically stay in the low “triple digits.”
If TMAO were the “killer,” fish-eaters should have the worst hearts on the planet. Instead, they have the best.
| Diet Type | Typical TMAO Level in Blood | Heart Health Outcome |
| High Red Meat Diet | Moderate Increase (Low Triple Digits) | Linked to higher heart risk in many studies |
| High Fish Diet | Massive Surge (Thousands of µmol/L) | Consistently linked to lower heart risk |
This paradox is the strongest evidence we have that TMAO is a “passenger,” not the “driver” of the car. It suggests that the molecule itself is likely harmless, but its presence in certain contexts tells us something is wrong elsewhere.
3. The Kidney Connection (The “Broken Filter” Problem)
In a healthy body, TMAO is a temporary guest. Once the liver refines it, the kidneys are supposed to flush it out into your urine. Scientists measure the “kidney’s cleaning speed” using a number called eGFR.
Think of your blood like a sink and your kidneys like the drain. If the drain is wide open, TMAO flows out easily. But if the drain gets even slightly clogged (low eGFR), TMAO begins to back up. This creates a massive scientific “confounder.” When a doctor finds high TMAO in a patient who just had a heart attack, was the TMAO the cause? Or did the patient have a “slow drain” (kidney trouble) that allowed the TMAO to pile up?
A landmark study of 1,159 individuals found that while high TMAO was linked to heart deaths, that link completely vanished once researchers accounted for the “kidney’s cleaning speed.”
If the “risk” disappears the moment you check the filter, it suggests TMAO isn’t the criminal—it’s just a sign that the plumbing is failing.
4. Your DNA Might Hold the Alibi
To solve the mystery of whether TMAO is a killer, scientists use a method called “Mendelian Randomization.” Think of this as “Nature’s Clinical Trial.” Since we are born with certain genes, some people naturally have higher TMAO levels regardless of what they eat.
If TMAO were a direct killer, these “genetically high” people should be having heart attacks left and right. But they aren’t. Large-scale genetic studies show that being born with “high TMAO genes” does not increase your risk of heart disease or stroke.
Fact Check:
The Cart Before the Horse Finding firefighters at a house doesn’t mean the firefighters caused the fire; it means the fire called the firefighters. Similarly, genetic data shows that people with diabetes or kidney disease naturally develop high TMAO. This is “Reverse Causation”: the disease causes the high TMAO, not the other way around.
5. When the Liver’s “Brakes” Fail
If TMAO is just a witness, why is it so high in people with weight gain and diabetes? The answer lies in the liver’s “refining machine,” the FMO3 enzyme.
In a healthy person, the hormone insulin acts as a brake on this machine. It tells the liver, “Slow down, we don’t need to churn out much TMAO right now.” But in people with “insulin resistance” (where the body’s cells stop listening to insulin), the brake fails.
The result is a runaway train. In a famous study of “LIRKO mice” (mice whose livers couldn’t hear the insulin signal), the genetic instructions for the TMAO machine (Fmo3 mRNA) increased by over 1,000-fold.
Crucial Note: While the instructions for the machine went up 1,000 times, the actual amount of TMAO in the blood only increased by 2.5 times. This shows that the body tries to manage the surge, but even a small 2.5-fold increase is a loud, clear signal that the liver’s “insulin brakes” are broken. High TMAO is the smoke; insulin resistance is the fire.
6. It’s the “Whole Neighborhood,” Not Just One Bad Actor
When we blame red meat for heart disease, we often get “tunnel vision” on TMAO. But red meat is a “bad neighborhood” filled with many potential suspects. TMAO is just one neighbor who happens to live there.
According to the latest research, red meat contains several components that might be working together to damage our pipes:
- Saturated Fats: These can “clog” the receptors in your liver, raising your “bad” cholesterol.
- Heme Iron: This can cause “chemical rusting” (called Fenton chemistry) in the arteries, damaging the lining.
- Sodium: Especially in processed meats like bacon, this increases blood pressure and stiffens arteries.
- Advanced Glycation End-products (AGEs): These are “inflammation triggers” formed during high-heat cooking. They engage something called RAGE, which essentially flips an “inflammation switch” in your blood vessels.
Even the famous “SWAP-MEAT” trial, which showed that people who switched to plant-based meat had lower TMAO, has a catch. The study was funded by a plant-based meat company, and more importantly, there was a major “order effect.” The TMAO reduction only happened for the people who ate animal meat first. If you started with plant meat, the effect wasn’t nearly as clear. This suggests that the “whole neighborhood” of your diet—how much fiber, fat, and sodium you eat—matters much more than one single molecule.
7. The “Uremic Toxin” Transition: Dr. Jekyll and Mr. Hyde
Is TMAO ever the villain? The evidence suggests TMAO has a “dual nature.” Much like the classic story of Dr. Jekyll and Mr. Hyde, TMAO is a peaceful citizen in a healthy body but can turn into a monster when the kidneys fail.
In people with advanced kidney disease, TMAO levels don’t just “rise”—they reach “supraphysiological” levels. At these extreme concentrations, TMAO stops being a “passenger” and starts becoming an active “uremic toxin” that may cause its own damage.
| Renal Status | Kidney “Cleaning Speed” | Best Interpretation of TMAO |
| Normal | Fast/Healthy | A harmless “passenger” reflecting your recent meals. |
| Mild-Moderate | Slowing Down | A “reporter” showing that your filters are starting to fail. |
| Advanced CKD | Very Slow / Stopped | A “Uremic Toxin” that may actively damage the heart. |
8. The Final Verdict: Reporter or Criminal?
So, should you worry about the TMAO in your steak or eggs?
The current weight of evidence suggests that for most of us, TMAO is a world-class reporter. It is a highly sensitive “integrative marker” that gives us a status report on four things:
- Your Diet: Are you eating more meat or more plants?
- Your Gut: What kind of bacteria are living inside you?
- Your Metabolism: Is your liver’s “insulin brake” working?
- Your Kidneys: How fast are you filtering your blood?
While it’s a great tool for predicting who might be at risk, trying to “kill” the TMAO molecule with drugs might be like smashing a smoke detector because you don’t like the noise. The detector isn’t the problem—the fire is.
The big question: Should we spend our energy trying to lower one molecule, or should we focus on fixing the “factory”—our diet, our activity levels, and our metabolic health—that produces it? For now, the science suggests that if you take care of the factory, the “reporter” will take care of itself.
DEEP DIVE
Trimethylamine N-Oxide in Cardiovascular Disease
A Comprehensive Evidence-Based Review of Causality Versus Biomarker Association
Editorial note on this revision
This is the second revision. The first corrected accuracy problems found on cross-check against the primary literature—most consequentially, the previous opening statistic (“a meta-analysis of 44 cohorts comprising over 84,000 individuals… 29% higher CVD risk, 51% higher cardiovascular mortality, 30% higher all-cause mortality”), which could not be located in any published meta-analysis and had been attributed to a commercial testing website; it was replaced with verified figures from the three principal meta-analyses (Qi 2018; Schiattarella 2017; Heianza 2017). The reference list was rebuilt around primary, PubMed/PMC-indexed sources.
This revision additionally incorporates an external accuracy audit. Its strongest contributions—adopted here—are the recalibration of the Bradford Hill and GRADE judgments (separating the strength of the observational association from the weaker case for causation), the restoration of the landmark Tang 2013 NEJM cohort, and tighter epistemic wording distinguishing “no evidence for a large causal effect” from “proof of reverse causation.” A subsequent polish pass added a tiered evidence summary, a renal-function-stratified analysis, an explicit limitations and future-research section, and a balanced account of the divergence in expert interpretation, and completed the migration of every citation to primary, peer-reviewed sources.
Two audit recommendations were declined on verification against primary sources: the proposed change of the γ-butyrobetaine figure from ~1,000-fold to ~50-fold (the ~1,000-fold value is the figure reported by Koeth 2014, Cell Metab), and the request to soften the murine FMO3-knockdown result (“entirely prevents…” is the authors’ own wording in Miao 2015, Nat Commun). Several other audit hedges reflected statistics the audit did not retrieve but that were independently verified here (e.g., the LIRKO >1,000-fold/2.5-fold figures and the Jia 2019 MR effect sizes). Volume/page details in the reference list should still receive a final cross-check before submission.
Biosynthetic Pathways and Meta-Organismal Metabolism of TMAO
The production of circulating trimethylamine N-oxide (TMAO) in humans is a multi-organismal process requiring coordinated interaction between dietary intake, the gastrointestinal microbiome, and host hepatic enzymes [1,3]. The cascade begins with ingestion of quaternary amine precursors—primarily choline, phosphatidylcholine (lecithin), L-carnitine, and betaine [1]. These nutrients are abundant in animal-derived foods such as red meat, egg yolks, and full-fat dairy, although they also serve essential roles in plant cellular membranes [1,2].
Once these precursors enter the gastrointestinal tract, a fraction escapes proximal absorption and transits to the distal ileum and cecum, where anaerobic microbiota cleave the carbon–nitrogen bonds of the precursors, releasing the volatile gas trimethylamine (TMA) [1]. Choline is converted directly to TMA by the microbial choline TMA-lyase system, encoded by the cutC gene and activated by its partner cutD [1].
L-carnitine catabolism follows a more complex, multi-step route mediated by distinct bacterial intermediates [2,4]. L-carnitine is first converted by gut bacteria to γ-butyrobetaine (γBB); the canonical carnitine monooxygenase (cntA/B) is oxygen-requiring and is therefore unlikely to be the dominant route in the largely anoxic colonic lumen, where an anaerobic γBB pathway predominates [4,5]. In isotope-tracer and mouse-intestinal incubation studies, γBB is generated at a rate roughly 1,000-fold higher than direct TMA formation and is the major gut-microbial metabolite of dietary L-carnitine; it is either absorbed into the systemic circulation—where it can act as an independent proatherogenic intermediate—or further metabolized by distinct anaerobes through crotonobetaine to TMA [4,5].
Newly synthesized TMA is lipophilic and diffuses across the intestinal mucosa into the portal circulation, which delivers it to the liver [3]. In hepatocytes, TMA is oxidized to the odorless, water-soluble TMAO, a reaction catalyzed by the flavin-containing monooxygenase (FMO) family, principally the FMO3 isoform—the most abundant and active FMO in the adult human liver [6,7].
Under normal physiological conditions, systemic TMAO is distributed throughout the extracellular fluid and cleared chiefly by the kidneys, with glomerular filtration as the dominant elimination route [3,18]. Renal clearance is therefore the single most important determinant of steady-state plasma concentrations in individuals with preserved kidney function [18,19].
Hepatic FMO3, Insulin Resistance, and Metabolic Syndrome
FMO3 is a metabolic hub linking the gut microbiome, host lipid transport, and systemic energy homeostasis [7,8]. Hepatic Fmo3 expression is under hormonal control: in insulin-sensitive states, insulin suppresses Fmo3 transcription, whereas glucagon and glucocorticoids stimulate it [6,7].
With obesity, visceral adiposity, and hepatic insulin resistance, this suppression is lost [7,8]. Chronic free-fatty-acid oversupply drives diacylglycerol accumulation and PKC-epsilon activation, which impairs hepatic insulin-receptor signaling; because insulin can no longer restrain Fmo3, hepatic FMO3 expression and activity rise [7,8].
In liver-specific insulin receptor knockout (LIRKO) mice—a model of selective hepatic insulin resistance—TMAO was the most strongly upregulated of 175 profiled hepatic metabolites, Fmo3 was the second most highly upregulated transcript, with a greater-than-1,000-fold increase in hepatic Fmo3 mRNA accompanied by an approximately 2.5-fold increase in plasma TMAO relative to controls [7]. In primary hepatocytes, insulin repressed and glucagon elevated FMO3 expression, and hepatic FMO3 mRNA was higher in heavily obese, hyperglycemic patients than in leaner, normoglycemic controls [7,8].
This upregulation is not an inert marker. Antisense knockdown of Fmo3 in insulin-resistant mice suppresses forkhead box protein O1 (FoxO1)—a central node for hepatic gluconeogenesis and lipogenesis—and, in these models, prevents the development of hyperglycemia, hyperlipidemia, and atherosclerosis even on a high-fat diet [7,8]. Mechanistically, FMO3 influences glucose and lipid handling partly via reduced lipid-induced endoplasmic-reticulum stress and modulation of SREBP-2/FoxO1 signaling [7,8].
Consequently, in subjects with preserved renal function, plasma TMAO may partly track hepatic insulin sensitivity: when insulin resistance lifts the brake on Fmo3, conversion of gut-derived TMA to systemic TMAO can accelerate [7,8]. This mechanism is well supported in murine and hepatocyte systems, and human liver-biopsy data show higher FMO3 expression in obese, hyperglycemic individuals; even so, current human evidence is insufficient to treat circulating TMAO as a validated standalone biomarker of hepatic insulin resistance. The relationship is best stated as mechanistically plausible rather than clinically established [7,8].
Causality: Mendelian Randomization Versus Observational Data
Whether TMAO is a direct causal mediator of atherosclerotic cardiovascular disease (ASCVD) or a correlative biomarker of metabolic and renal dysfunction is best examined by comparing observational and genetic designs [13]. Observational cohorts show a consistent, dose-dependent association between elevated plasma TMAO and major adverse cardiovascular events (MACE) [10,11,12]. In the landmark prospective study of 4,007 patients undergoing elective coronary angiography, higher fasting TMAO predicted death, myocardial infarction, or stroke over three years, with a hazard ratio of 2.54 for the highest versus lowest quartile that remained significant after adjustment for traditional risk factors [3].
The principal published meta-analyses quantify this association. Qi et al. (2018), pooling 11 prospective cohorts, reported a 23% higher risk of cardiovascular events (HR 1.23, 95% CI 1.07–1.42) and a 55% higher risk of all-cause mortality (HR 1.55, 95% CI 1.19–2.02) for higher versus lower TMAO [10]. Schiattarella et al. (2017), pooling 17 studies enrolling 26,167 subjects, found high TMAO associated with a 91% higher all-cause mortality (HR 1.91, 95% CI 1.40–2.61) and a 67% higher risk of major adverse cardiovascular and cerebrovascular events (HR 1.67, 95% CI 1.33–2.11), with a dose-response of roughly a 7.6% increase in mortality risk per 10 µmol/L increment [11]. Heianza et al. (2017) similarly reported elevated TMAO associated with greater MACE (HR 1.41) and all-cause death (HR 1.55) [12]. These observational signals are echoed by experimental data: in animal and cellular models, TMAO or its precursors have been shown to promote foam-cell formation, upregulate macrophage scavenger receptors (CD36, SR-A), impair reverse cholesterol transport, and enhance platelet activation and thrombosis [2,4,21].
Large-scale Mendelian randomization (MR) challenges the causal interpretation [13]. Using single-nucleotide polymorphisms as instruments for lifetime exposure, a bidirectional two-sample MR drawing on DIAGRAM, CARDIoGRAMplusC4D, and the UK Biobank found that genetically predicted higher TMAO and L-carnitine were not associated with coronary artery disease, myocardial infarction, stroke, atrial fibrillation, type 2 diabetes, or chronic kidney disease after Bonferroni correction (P ≤ 0.0005) [13].
The same analysis revealed a reverse-causal direction: genetic liability to type 2 diabetes (β = 0.130 ± 0.036, P < 0.0001) and to chronic kidney disease (β = 0.483 ± 0.168, P = 0.004) was causally associated with higher circulating TMAO [13]. Read carefully, these findings do not establish a large direct causal effect of lifelong higher TMAO on cardiovascular disease; combined with the reverse-direction signals, they are consistent with the authors’ conclusion that much of the observational association may be explained by confounding and reverse causation—a more defensible statement than asserting that every observed association is reverse-caused [13].
A separate MR analysis did suggest a possible causal contribution of genetically predicted TMAO and L-carnitine to higher systolic blood pressure, indicating that while primary atherogenesis is not genetically supported, a minor role in vascular remodeling and hypertensive pathology cannot be excluded [14].
Systemic Integration of Confounding Factors
Several variables alter both circulating TMAO and cardiovascular risk and account for much of the cross-design discrepancy:
- Renal function (eGFR, cystatin C): glomerular filtration is the primary clearance route, so any reduction in eGFR raises systemic TMAO independent of diet or microbiome activity—making renal function the single largest confounder in observational TMAO studies [18,19].
- Insulin sensitivity and metabolic syndrome: hepatic insulin resistance upregulates FMO3, accelerating conversion of TMA to TMAO; circulating TMAO may therefore partly reflect hepatic insulin sensitivity, though human validation remains incomplete [7,8].
- Systemic inflammation: TMAO correlates with hsCRP and IL-6, which independently drive arterial remodeling and plaque instability [20].
- Advancing age: aging lowers GFR and hepatic perfusion and raises arterial stiffness, all of which correlate with higher plasma TMAO [9,31].
- Microbiome composition: the efficiency of precursor-to-TMA conversion depends on community structure (e.g., relative cutC/cutD and cntA/B carriage), so taxonomic differences shift systemic TMAO [1,4].
- Medications and xenobiotics: broad-spectrum antibiotics suppress gut microbiota and abolish TMAO production, but there is no evidence that antibiotic suppression of TMAO translates into improved long-term cardiovascular outcomes (no such trial has been designed to test this); conversely, standard heart-failure therapies improve myocardial strain markers without lowering TMAO, indicating these agents do not act through the TMAO pathway [3,21].
Independent Predictive Value and the Renal Confounder
A central question in clinical lipidology is whether TMAO predicts events after adjustment for renal function and traditional risk factors [16]. Because TMAO is renally cleared, its prognostic significance depends heavily on how kidney function is handled in multivariable models [16,17].
When cohorts adjust for baseline eGFR or cystatin C, the prognostic signal of TMAO is frequently attenuated or abolished. In a large Danish cohort of approximately 1,159 individuals with type 1 diabetes followed for a median of about 15 years, higher plasma TMAO was associated with all-cause and cardiovascular mortality, combined CVD events, coronary outcomes, stroke, heart-failure hospitalization, and end-stage renal disease, independent of conventional risk factors; after further adjustment for baseline eGFR, the associations became non-significant across all endpoints, and TMAO was strongly inversely related to baseline eGFR (R² = 0.29, P < 0.001)—consistent with its prognostic value being largely a function of renal clearance [15].
The PREVEND general-population cohort shows the same pattern: among 5,469 participants over a median 8.3 years, TMAO predicted all-cause mortality after risk-factor adjustment (HR 1.36, 95% CI 0.97–1.91), but the association was lost after further adjustment for albuminuria and eGFR (HR 1.15, 95% CI 0.81–1.64), and the TMAO–mortality relationship was significantly modified by renal function (interaction P = 0.002) [17].
Community-based cohorts of older adults reinforce this. In the Cardiovascular Health Study, serial TMAO predicted incident and recurrent ASCVD, but further adjustment for eGFR attenuated the recurrent-ASCVD association to non-significance (HR 1.10, 95% CI 0.87–1.39; P-trend = 0.179), and the TMAO signal was concentrated in participants with reduced renal function [16]. The companion Multi-Ethnic Study of Atherosclerosis (6,767 adults) likewise found a dose-dependent TMAO–ASCVD association (quintile HRs 1.02, 1.17, 1.23, 1.33) that appeared stronger in those with baseline eGFR below 60 mL/min/1.73 m² [16].
These observations support a dual-nature interpretation. In normal renal function and preserved insulin sensitivity, physiological fluctuations in TMAO appear clinically benign—they have not been shown to increase cardiovascular events—and the biomarker behaves as a passenger [8]. In moderate-to-severe CKD, however, a vicious uremic cycle—dysbiosis, increased mucosal permeability, and impaired clearance—produces supraphysiological accumulation [18,19]. At those concentrations, experimental work links TMAO to NLRP3 inflammasome activation, TGF-β/Smad3-driven tubulointerstitial fibrosis, and endothelial injury, suggesting a transition from passive marker toward active uremic toxin in advanced kidney disease [19,30].
The strength of the evidence for TMAO pathogenicity therefore varies markedly across the spectrum of renal function, and the two are best kept distinct. With normal renal function, TMAO is chiefly a diet- and microbiome-responsive marker with no demonstrated independent event risk; in mild-to-moderate CKD, concentrations rise as clearance falls and the prognostic association strengthens but remains difficult to disentangle from declining eGFR itself; in advanced CKD and on dialysis, TMAO reaches severalfold-higher concentrations, tracks renal function tightly, and—supported by experimental cardiorenal models—is most plausibly a contributor to, not merely a marker of, cardiorenal pathology [17,18,19,31].
Table 5. TMAO across the spectrum of renal function.
| Renal status | Typical TMAO | Prognostic association | Best interpretation |
| Normal (eGFR ≥ 60, no albuminuria) | Low, diet-responsive | Weak/absent after adjustment | Passenger marker of diet and microbiome [16] |
| Mild–moderate CKD | Progressively elevated | Present but eGFR-entangled | Marker of clearance and disease severity [16,17] |
| Advanced CKD / dialysis | Severalfold elevated | Strong; tracks renal function | Plausible active contributor to cardiorenal injury [18,19,31] |
Dietary Patterns and Post-Challenge Metabolic Kinetics
Long-term diet shapes the capacity to synthesize TMAO. In carnitine- or choline-challenge studies, omnivores generate substantially more TMAO post-ingestion than long-standing vegetarians and vegans, a difference driven by chronic exposure to animal-derived nutrients selecting for TMA-producing taxa [5,24]. Suppressing the microbiota with broad-spectrum antibiotics abolishes post-challenge TMAO in both groups, confirming the obligatory microbial step [5].
This capacity is reduced but not absent in plant-based eaters: long-term vegetarians can still mount substantial TMAO responses to an oral carnitine challenge [24]. In a double-blind randomized pilot, a single lean-vegan-donor fecal microbiota transplant into omnivores with metabolic syndrome shifted recipient microbiota composition but did not alter carnitine- or choline-to-TMAO conversion or arterial-wall inflammation over two weeks, underscoring the resilience of the host metabolic phenotype [23].
Table 1. Dietary pattern and TMAO handling (directional summary).
| Dietary profile | Fasting plasma TMAO | Post-challenge kinetics (oral carnitine/choline) | Dominant microbiota features | CVD event risk |
| Vegans | Low [24] | Flat / minimal TMAO or γBB generation [5,24] | High fiber-fermenting taxa; low cutC/cutD carriage | Lower in observational dietary studies (a dietary-pattern, not a TMAO-specific, finding) [23,24] |
| Vegetarians | Low–moderate [24] | Intermediate; residual conversion capacity retained [24] | High diversity; variable cutC/cutD and cntA/B | Reduced vs typical omnivores [24] |
| Omnivores | Moderate [5] | Robust, rapid post-prandial rise in TMAO and γBB [5] | Higher carriage of carnitine/choline-converting taxa | Standard reference [16] |
| High red-meat consumers | Elevated [2,5] | Large, prolonged post-prandial surges of TMAO and γBB [5] | Enriched carnitine-utilizing pathway [5] | Elevated (matrix-dependent) [16,25] |
Plant-rich diets keep baseline and post-prandial TMAO low partly through soluble fiber and polyphenols: fiber acts as a prebiotic favoring SCFA-producing taxa and slowing nutrient absorption, while polyphenols appear capable of inhibiting microbial TMA-lyase activity in experimental models [5,24]. Whether lower TMAO is itself the mechanism for the reduced cardiovascular risk of plant-based diets is unresolved, because such diets simultaneously lower LDL-C, blood pressure, hsCRP, and body weight—making the independent contribution of TMAO reduction difficult to isolate [10,11].
The Fish Consumption Paradox
The strongest empirical challenge to a direct-toxin model is the fish paradox [20]. Marine fish accumulate high concentrations of preformed, free TMAO as an osmolyte that stabilizes proteins against pressure, salinity, and urea [20]. Because preformed TMAO is absorbed directly without a microbial step, seafood produces rapid, large spikes in plasma and urinary TMAO [3].
The magnitude is striking. A cod or halibut meal can raise plasma TMAO several-fold within hours—for example, from a baseline near 9 µmol/L to roughly 23 µmol/L after codfish in one controlled challenge [27]—and older dietary studies reported that habitual seafood consumers reach far higher levels than those on egg-and-red-meat diets (on the order of thousands of µmol/L versus low triple digits in the cited pilot data) [28]. These excursions exceed those typically achieved on a high-red-meat or egg diet [27,28].
If circulating TMAO were a direct driver of atherosclerosis, thrombosis, and arterial inflammation, fish-rich diets should accelerate disease; instead, fish and seafood consumption is consistently associated with reduced coronary heart disease, stroke, sudden cardiac death, and all-cause mortality, and the benefit extends to lean white fish with negligible omega-3 content [20]. Several arguments attempt to reconcile the paradox, each with limitations:
- Marine-lipid masking: EPA/DHA anti-inflammatory and anti-arrhythmic effects override TMAO toxicity—but this fails to explain the benefit of lean white fish low in omega-3 [20].
- Transient kinetics: seafood-induced spikes are cleared within ~24 h and so are benign, whereas microbial synthesis from red meat is chronic—but habitual seafood eaters maintain chronically elevated baseline TMAO without evident vascular harm [20].
- Dysbiosis-marker hypothesis: TMA-derived TMAO is harmful chiefly as a surrogate for a dysbiotic, pro-inflammatory gut microbiome and impaired barrier, whereas seafood-derived TMAO bypasses that microbial step and neither reflects nor induces dysbiosis [20].
The fish-feeding literature materially weakens a simple monotonic “higher TMAO equals greater vascular toxicity” model. It does not by itself exclude context-specific effects—for example in acute thrombosis or advanced CKD—but it weighs strongly against circulating TMAO acting as a primary vascular toxin at physiological or moderately elevated human concentrations [20].
Red Meat and Cardiovascular Risk: Disentangling TMAO From Alternative Mechanisms
Atherosclerosis is driven by subendothelial retention of apolipoprotein-B lipoproteins and recruitment of monocyte-derived foam cells. Whether unprocessed red meat accelerates this chiefly through the TMAO pathway or via other mechanisms is unsettled, because red meat carries several bioactive components, each independently linked to vascular injury [25].
Table 2. Candidate atherogenic mechanisms of red-meat components.
| Component | Proposed mechanism | Associated biomarkers | Principal modifiers |
| Saturated fatty acids | Downregulate hepatic LDL receptors, raising LDL-C/ApoB and arterial-wall retention | LDL-C, ApoB | Replacing SFA with PUFA or plant protein lowers risk [25] |
| Heme iron | Catalyzes ROS via Fenton chemistry, promoting LDL peroxidation | Oxidized LDL, MDA, F2-isoprostanes | Calcium and polyphenols chelate iron, reducing oxidation |
| Sodium (processed meats) | Endothelial shear stress, arterial stiffness, volume-dependent hypertension | Blood pressure, NT-proBNP | Potassium-rich (DASH) patterns mitigate |
| Advanced glycation end-products | RAGE engagement activates NF-κB and vascular adhesion molecules | hsCRP, TNF-α, IL-6 | Low-temperature cooking reduces AGE formation |
| L-carnitine / TMAO pathway | Suppresses reverse cholesterol transport; upregulates scavenger receptors | TMAO, γBB, crotonobetaine | Fiber- and polyphenol-rich (Mediterranean) matrix attenuates [5,24] |
Although rodent feeding of L-carnitine or choline raises atherosclerosis in a microbiota-dependent manner [2], isolating this effect in humans is difficult [25]. In randomized trials, the cardiovascular risk of unprocessed red meat depends strongly on the comparison diet: replacing red meat with high-quality plant protein lowers LDL-C and coronary heart disease incidence [25]. Notably, incorporating lean unprocessed beef within a Mediterranean-style pattern (rich in monounsaturated fat, fiber, and polyphenols) can increase microbiota diversity and lower circulating and urinary TMAO relative to a high-saturated-fat American diet [25].
This indicates the proatherogenic potential of red meat is not driven by L-carnitine or TMAO generation in isolation, but by the broader dietary matrix—high saturated fat, low fiber, high sodium, and inflammation interacting with microbial pathways [5,25]. The SWAP-MEAT crossover trial (n = 36; single-site, no washout period) illustrates the interpretive caution required: overall TMAO was lower during the plant-based-meat phase (2.7 vs 4.7 µmol/L, P = 0.012), but the trial was funded by a plant-based-meat manufacturer, and the effect was driven by a significant order effect (P = 0.023)—the difference appeared only in participants who consumed animal meat first (2.9 vs 6.4 µmol/L, P = 0.007) and not in those who consumed plant-based meat first (2.5 vs 3.0 µmol/L, P = 0.23). The trial did not—and with n = 36 was underpowered to—show that these transient TMAO shifts translate into differences in arterial inflammation or hard clinical events [22].
Systematic Evaluation Using the Bradford Hill Criteria
Table 3. Bradford Hill appraisal of circulating TMAO as a causal mediator of CVD.
| Criterion | Application to TMAO and cardiovascular disease | Status |
| Strength | Observational HRs of roughly 1.2–2.5 for events and mortality (e.g., NEJM HR 2.54 in a high-risk angiography cohort); modest in lower-risk settings and markedly attenuated after eGFR adjustment [3,10,11,16,17] | Weak–moderate |
| Consistency | Highly reproducible as an observational association across cohorts, but not corroborated as a causal effect in Mendelian randomization [3,10,11,12,13] | High for association; low across designs |
| Specificity | Elevated TMAO accompanies diabetes, NAFLD, obesity, CKD, and other conditions; not specific to vascular disease [7,18] | Unmet |
| Temporality | Prospective cohorts establish that elevated TMAO precedes events (temporal ordering met); bidirectional MR indicates this ordering is confounded by reverse causation from T2DM and CKD [3,13] | Met for ordering; confounded |
| Biological gradient | Dose-response reported for mortality and for blood pressure, but remains vulnerable to renal-function confounding [11,14] | Partially met |
| Plausibility | Coherent cellular pathways: scavenger-receptor upregulation, impaired reverse cholesterol transport, NF-κB/NLRP3 activation, platelet hyperreactivity [4,21] | Met (experimental) |
| Coherence | The fish paradox conflicts with a simple toxin model—seafood spikes TMAO yet is cardioprotective—though context-specific effects are not excluded [20] | Partially unmet |
| Experiment | Rodent feeding accelerates atherosclerosis; no human trial yet shows that lowering TMAO reduces events or vascular inflammation [2,29] | Partial (animal only) |
| Analogy | Resembles clearance-dependent markers (ADMA, cystatin C) reflecting kidney-heart crosstalk; analogy is hypothesis-generating, not causal evidence [15,17] | Weak / not probative |
On balance, the observational association is strong and biologically plausible, but the criteria that bear most on causation—cross-design consistency, specificity, freedom from reverse-causation confounding, and coherence with the fish data—are only partially met or unmet. The framework therefore supports TMAO as a robust risk marker while falling short of establishing it as a primary causal driver of human ASCVD [13,20].
GRADE Certainty of Evidence
GRADE is best applied to discrete questions rather than to a single pooled judgment that mixes prognosis, genetics, animal mechanism, and diet responsiveness. The evidence is therefore separated into four distinct questions, each with its own certainty rating.
Table 4. GRADE summary by question.
| Question | Finding | Certainty | Basis |
| Is TMAO a prognostic biomarker in high-risk cohorts? | Baseline TMAO predicts MACE, heart failure, and mortality in CAD, ACS, HF, and CKD populations [3,10,11,12] | Moderate | Large consistent cohorts with dose-response, downgraded for substantial confounding by renal function and disease severity [15,16,17] |
| Does TMAO causally drive human ASCVD events? | Genetically predicted TMAO/carnitine show no direct link to CAD, MI, stroke, AF, or T2DM; rodent feeding accelerates plaque [2,13] | Low | Animal plausibility but no MR support for a large direct effect; rodent indirectness (no CETP, different bile-acid handling, supraphysiological doses) [7,13] |
| Does lowering TMAO improve clinical outcomes? | TMA-lyase inhibitors lower TMAO and atherosclerosis in animals; no human outcome trial exists [21,29] | Very low | No randomized human hard-outcome trial of selective TMAO lowering has been reported |
| Can diet meaningfully change circulating TMAO? | Red meat raises, and fiber-/plant-rich diets and antibiotic suppression lower, circulating TMAO [5,25] | High | Consistent high-quality randomized crossover and challenge studies showing rapid microbial plasticity [5,25] |
Current state of the evidence at a glance
Synthesizing the above, the individual claims commonly grouped under the “TMAO hypothesis” sit at very different levels of certainty and should not be asserted with uniform confidence.
Table 6. Tiered summary of TMAO evidence, strongest to weakest.
| Certainty tier | Claims supported at this level |
| Well established (high certainty) | TMAO predicts cardiovascular risk in high-risk cohorts; reduced renal function raises circulating TMAO; seafood raises TMAO; gut microbiota generate TMA from dietary precursors; diet meaningfully modifies TMAO |
| Moderate certainty | TMAO/FMO3 biology is linked to metabolic dysfunction; animal and cellular mechanisms for atherogenesis, thrombosis, and cholesterol handling are biologically plausible; a modest causal effect on systolic blood pressure is possible |
| Weak or conflicting | Circulating TMAO directly causes human atherosclerotic events; lowering TMAO reduces cardiovascular events; a defined human “toxic threshold” exists outside advanced CKD |
Scientific Consensus and Remaining Controversies
Consensus has shifted from viewing TMAO as a simple vascular toxin to recognizing it as an integrative metabolic reporter sitting at the intersection of diet quality, microbiome composition, hepatic metabolism, and renal clearance [10,16]. As a prognostic tool, elevated TMAO usefully stratifies risk—particularly in established CAD, ACS, heart failure, and CKD—capturing kidney-heart-gut dysfunction not reflected in standard lipid, glycemic, or inflammatory panels [16,18,32]. Several controversies persist:
- Causality: despite plausibility and rodent pathogenicity, the absence of MR support plus the fish paradox lead many investigators to regard circulating TMAO as primarily a passenger at normal human concentrations [13,20].
- Therapeutic targeting: non-lethal TMA-lyase inhibitors (e.g., 3,3-dimethyl-1-butanol, iodomethylcholine) reduce TMAO, platelet hyperreactivity, and atherosclerosis in animals, but whether they lower human events is unknown; critics argue that suppressing a downstream marker without addressing diet, inactivity, and adiposity may not reduce events [29].
- The eGFR-adjustment question: some hold that adjusting for eGFR over-corrects because TMAO may itself contribute to renal decline (i.e., a mediator), while others insist that without rigorous adjustment for measured GFR or cystatin C the observational signal is confounded by subclinical kidney impairment [16,17].
These controversies map onto a genuine divergence in expert interpretation. The causal hypothesis has been advanced most vigorously by the Cleveland Clinic group whose experimental program produced much of the foundational work—the original phosphatidylcholine and L-carnitine pathway studies, the FMO3 and platelet-hyperreactivity mechanisms, and the TMA-lyase inhibitor proof-of-concept [1,2,4,21,29]—and which reads the convergent cohort and mechanistic data as evidence of a modifiable causal contributor. A more cautious reading, prominent among cardiovascular epidemiologists and nutrition researchers, emphasizes the null Mendelian randomization findings, the dominant role of renal clearance as a confounder, and the fish paradox, and treats TMAO as chiefly a marker of diet, microbiome, and kidney-heart crosstalk. The two positions are not as far apart as they appear: both accept the prognostic value and the microbial pathway, and disagree mainly on whether lowering circulating TMAO would alter human events—an empirical question that only a randomized outcome trial can settle [13,20,29].
In sum, TMAO remains a valuable biomarker for risk stratification and precision nutrition, but the current weight of genetic and epidemiological evidence argues against a primary causal role in human atherosclerotic disease [13]. Randomized trials of selective TMA-lyase inhibition will be needed to determine whether lowering circulating TMAO reduces events—assuming an agent proves safe for long-term human use, since all successful work to date remains preclinical—or whether clinical focus should remain on the fundamental drivers: diet quality, physical activity, insulin sensitivity, and renal health [13,29].
Limitations of the Current Evidence Base
Several constraints bound every conclusion above and are stated explicitly here rather than left implicit.
- Most human evidence is observational, and even large, well-adjusted cohorts cannot fully exclude residual confounding by renal function, metabolic disease severity, and diet quality.
- Human intervention trials that lower TMAO and measure hard cardiovascular endpoints do not exist; the therapeutic question is therefore unresolved rather than settled negatively.
- Mendelian randomization has its own limitations—potential pleiotropy of metabolite-associated variants, instruments derived from relatively small exposure GWAS, and assumptions that cannot be fully verified—so null MR results argue against a large lifelong effect but do not exclude context-specific or acute effects.
- Animal and cellular models frequently require supraphysiological exposures and differ from humans in lipoprotein handling (e.g., absence of CETP) and bile-acid metabolism, limiting direct translation.
- TMAO assays and reference ranges are not fully standardized across laboratories, complicating cross-study comparison of absolute concentrations and any proposed thresholds.
- TMAO may exert different biological effects depending on concentration and clinical context—plausibly inert at physiological levels yet pathogenic at the supraphysiological concentrations of advanced CKD—so a single verdict across all populations is inappropriate.
Future Research Directions
The following questions would most efficiently move the field from association toward causal clarity:
- Do selective TMA-lyase inhibitors reduce major adverse cardiovascular events in adequately powered, long-term randomized human trials, and are they safe for chronic use?
- Is there a definable TMAO concentration threshold above which pathogenicity emerges, particularly across CKD stages?
- Are TMA and TMAO biologically distinct in their vascular and renal effects, and which is the more proximate mediator?
- Do common FMO3 genetic variants alter cardiovascular or renal risk in humans, providing a natural experiment for causal inference?
- Does TMAO have qualitatively different effects in advanced CKD versus preserved renal function, as the dual-nature model predicts?
- Can durable gut-microbiome manipulation (diet, prebiotics, or engineered consortia) sustainably lower TMAO, and does doing so improve vascular endpoints independent of concomitant LDL-C, blood-pressure, and weight changes?
References
- Wang Z, Klipfell E, Bennett BJ, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472(7341):57-63. doi:10.1038/nature09922
- Koeth RA, Wang Z, Levison BS, et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013;19(5):576-585. doi:10.1038/nm.3145
- Tang WH, Wang Z, Levison BS, et al. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med. 2013;368(17):1575-1584. doi:10.1056/NEJMoa1109400
- Koeth RA, Levison BS, Culley MK, et al. γ-Butyrobetaine is a proatherogenic intermediate in gut microbial metabolism of L-carnitine to TMAO. Cell Metab. 2014;20(5):799-812. doi:10.1016/j.cmet.2014.10.006
- Koeth RA, Lam-Galvez BR, Kirsop J, et al. l-Carnitine in omnivorous diets induces an atherogenic gut microbial pathway in humans. J Clin Invest. 2019;129(1):373-387. doi:10.1172/JCI94601
- Bennett BJ, de Aguiar Vallim TQ, Wang Z, et al. Trimethylamine-N-oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation. Cell Metab. 2013;17(1):49-60. doi:10.1016/j.cmet.2012.12.011
- Miao J, Ling AV, Manthena PV, et al. Flavin-containing monooxygenase 3 as a potential player in diabetes-associated atherosclerosis. Nat Commun. 2015;6:6498. Published 2015 Apr 7. doi:10.1038/ncomms7498
- Shih DM, Wang Z, Lee R, et al. Flavin containing monooxygenase 3 exerts broad effects on glucose and lipid metabolism and atherosclerosis. J Lipid Res. 2015;56(1):22-37. doi:10.1194/jlr.M051680
- Schugar RC, Shih DM, Warrier M, et al. The TMAO-Producing Enzyme Flavin-Containing Monooxygenase 3 Regulates Obesity and the Beiging of White Adipose Tissue. Cell Rep. 2017;19(12):2451-2461. doi:10.1016/j.celrep.2017.05.077
- Qi J, You T, Li J, et al. Circulating trimethylamine N-oxide and the risk of cardiovascular diseases: a systematic review and meta-analysis of 11 prospective cohort studies. J Cell Mol Med. 2018;22(1):185-194. doi:10.1111/jcmm.13307
- Schiattarella GG, Sannino A, Toscano E, et al. Gut microbe-generated metabolite trimethylamine-N-oxide as cardiovascular risk biomarker: a systematic review and dose-response meta-analysis. Eur Heart J. 2017;38(39):2948-2956. doi:10.1093/eurheartj/ehx342
- Heianza Y, Ma W, Manson JE, Rexrode KM, Qi L. Gut Microbiota Metabolites and Risk of Major Adverse Cardiovascular Disease Events and Death: A Systematic Review and Meta-Analysis of Prospective Studies. J Am Heart Assoc. 2017;6(7):e004947. Published 2017 Jun 29. doi:10.1161/JAHA.116.004947
- Jia J, Dou P, Gao M, et al. Assessment of Causal Direction Between Gut Microbiota-Dependent Metabolites and Cardiometabolic Health: A Bidirectional Mendelian Randomization Analysis. Diabetes. 2019;68(9):1747-1755. doi:10.2337/db19-0153
- Wang H, Luo Q, Ding X, Chen L, Zhang Z. Trimethylamine N-oxide and its precursors in relation to blood pressure: A mendelian randomization study. Front Cardiovasc Med. 2022;9:922441. Published 2022 Jul 22. doi:10.3389/fcvm.2022.922441
- Winther SA, Øllgaard JC, Tofte N, et al. Utility of Plasma Concentration of Trimethylamine N-Oxide in Predicting Cardiovascular and Renal Complications in Individuals With Type 1 Diabetes. Diabetes Care. 2019;42(8):1512-1520. doi:10.2337/dc19-0048
- Lee Y, Nemet I, Wang Z, et al. Longitudinal Plasma Measures of Trimethylamine N-Oxide and Risk of Atherosclerotic Cardiovascular Disease Events in Community-Based Older Adults. J Am Heart Assoc. 2021;10(17):e020646. doi:10.1161/JAHA.120.020646
- Gruppen EG, Garcia E, Connelly MA, et al. TMAO is Associated with Mortality: Impact of Modestly Impaired Renal Function. Sci Rep. 2017;7(1):13781. Published 2017 Oct 23. doi:10.1038/s41598-017-13739-9
- Stubbs JR, House JA, Ocque AJ, et al. Serum Trimethylamine-N-Oxide is Elevated in CKD and Correlates with Coronary Atherosclerosis Burden. J Am Soc Nephrol. 2016;27(1):305-313. doi:10.1681/ASN.2014111063
- Tang WH, Wang Z, Kennedy DJ, et al. Gut microbiota-dependent trimethylamine N-oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ Res. 2015;116(3):448-455. doi:10.1161/CIRCRESAHA.116.305360
- Velasquez MT, Ramezani A, Manal A, Raj DS. Trimethylamine N-Oxide: The Good, the Bad and the Unknown. Toxins (Basel). 2016;8(11):326. Published 2016 Nov 8. doi:10.3390/toxins8110326
- Zhu W, Gregory JC, Org E, et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell. 2016;165(1):111-124. doi:10.1016/j.cell.2016.02.011
- Crimarco A, Springfield S, Petlura C, et al. A randomized crossover trial on the effect of plant-based compared with animal-based meat on trimethylamine-N-oxide and cardiovascular disease risk factors in generally healthy adults: Study With Appetizing Plantfood-Meat Eating Alternative Trial (SWAP-MEAT). Am J Clin Nutr. 2020;112(5):1188-1199. doi:10.1093/ajcn/nqaa203
- Smits LP, Kootte RS, Levin E, et al. Effect of Vegan Fecal Microbiota Transplantation on Carnitine- and Choline-Derived Trimethylamine-N-Oxide Production and Vascular Inflammation in Patients With Metabolic Syndrome. J Am Heart Assoc. 2018;7(7):e008342. Published 2018 Mar 26. doi:10.1161/JAHA.117.008342
- Wu WK, Chen CC, Liu PY, et al. Identification of TMAO-producer phenotype and host-diet-gut dysbiosis by carnitine challenge test in human and germ-free mice. Gut. 2019;68(8):1439-1449. doi:10.1136/gutjnl-2018-317155
- Guasch-Ferré M, Satija A, Blondin SA, et al. Meta-Analysis of Randomized Controlled Trials of Red Meat Consumption in Comparison With Various Comparison Diets on Cardiovascular Risk Factors. Circulation. 2019;139(15):1828-1845. doi:10.1161/CIRCULATIONAHA.118.035225
- O’Connor LE, Kim JE, Clark CM, Zhu W, Campbell WW. Effects of Total Red Meat Intake on Glycemic Control and Inflammatory Biomarkers: A Meta-Analysis of Randomized Controlled Trials. Adv Nutr. 2021;12(1):115-127. doi:10.1093/advances/nmaa096
- Regis B, Passeri L, Moreira NX, Borges NA, Ribeiro-Alves M, Mafra D. Plasma Trimethylamine N-Oxide Levels in Nondialysis Chronic Kidney Disease Patients Following Meal Challenge. Mol Nutr Food Res. 2025;69(14):e70121. doi:10.1002/mnfr.70121
- Zhang AQ, Mitchell SC, Smith RL. Dietary precursors of trimethylamine in man: a pilot study. Food Chem Toxicol. 1999;37(5):515-520. doi:10.1016/s0278-6915(99)00028-9
- Wang Z, Roberts AB, Buffa JA, et al. Non-lethal Inhibition of Gut Microbial Trimethylamine Production for the Treatment of Atherosclerosis. Cell. 2015;163(7):1585-1595. doi:10.1016/j.cell.2015.11.055
- Li Y, Lu H, Guo J, et al. Gut microbiota-derived trimethylamine N-oxide is associated with the risk of all-cause and cardiovascular mortality in patients with chronic kidney disease: a systematic review and dose-response meta-analysis. Ann Med. 2023;55(1):2215542. doi:10.1080/07853890.2023.2215542
- Chen G, He L, Dou X, Liu T. Association of Trimethylamine-N-Oxide Levels with Risk of Cardiovascular Disease and Mortality among Elderly Subjects: A Systematic Review and Meta-Analysis. Cardiorenal Med. 2022;12(2):39-54. doi:10.1159/000520910
- Senthong V, Wang Z, Li XS, et al. Intestinal Microbiota-Generated Metabolite Trimethylamine-N-Oxide and 5-Year Mortality Risk in Stable Coronary Artery Disease: The Contributory Role of Intestinal Microbiota in a COURAGE-Like Patient Cohort. J Am Heart Assoc. 2016;5(6):e002816. Published 2016 Jun 10. doi:10.1161/JAHA.115.002816