
ApoB, Insulin Resistance, and Cardiovascular Risk
A Research Analysis of Insulin Resistance, Apolipoprotein B (ApoB), and Cardiovascular Risk
1. ApoB as a Causal Cardiovascular Risk Factor
Atherosclerotic cardiovascular disease (ASCVD) is a primary global threat to human health, driven largely by dyslipidemia.[1] Traditionally, clinical assessment of lipid-associated risk has relied on low-density lipoprotein cholesterol (LDL-C) mass.[1] However, a more comprehensive physiological understanding reveals that apolipoprotein B-100 (ApoB) provides a superior and more direct representation of atherogenic particle burden.[3]
Biology of ApoB-Containing Lipoproteins
Every atherogenic lipoprotein particle synthesized by the liver carries exactly one molecule of ApoB-100 on its surface.[4] These particles span a continuous spectrum of density and size, including:
- Very-low-density lipoproteins (VLDL): large, triglyceride-rich particles secreted by the liver that deliver fatty acids to peripheral tissues.[4]
- Intermediate-density lipoproteins (IDL): transitional particles formed during the lipolysis of VLDL.[4]
- Low-density lipoproteins (LDL): the final, cholesterol-dense remnants of VLDL lipolysis and the primary carriers of circulating cholesterol.[4]
- Remnant particles: partially lipolyzed VLDL and chylomicrons (the latter containing ApoB-48) that are highly atherogenic.[6]
- Lipoprotein(a) [Lp(a)]: an LDL-like particle with an additional apolipoprotein(a) moiety covalently bound to the ApoB molecule.[5]
Because of this rigid stoichiometric relationship, measuring the total serum concentration of ApoB provides an exact count of all circulating atherogenic particles, irrespective of their lipid cargo.[4]
ApoB vs. LDL-C: Particle Count vs. Cholesterol Mass
LDL-C measures the total mass of cholesterol contained within LDL particles, rather than the concentration of the particles themselves.[3] However, the cholesterol content per particle is highly variable, influenced by systemic metabolic conditions and lipid remodeling.[4] In patients with metabolic dysfunction, high triglycerides, or insulin resistance, LDL particles frequently undergo depletion of their cholesterol core, remodeling into smaller, denser particles.[9] Consequently, an individual may have a normal or even low LDL-C mass while harboring a highly elevated number of small, dense LDL particles.[9]
Under such discordant scenarios, LDL-C systematically underestimates the true atherogenic burden, whereas ApoB correctly quantifies the absolute particle count.[1]
This does not make ApoB universally superior in every setting. Non-HDL-C is an inexpensive, validated, and widely guideline-endorsed alternative that captures all ApoB-containing lipoproteins without an additional assay, and several analyses find that ApoB, non-HDL-C, and LDL particle number perform similarly when lipid levels are concordant; ApoB’s incremental value is greatest precisely in the discordant, insulin-resistant phenotype that is the focus of this review. The balance of expert opinion increasingly favors ApoB as the single most direct measure of atherogenic-particle number, but reasonable debate remains about its incremental value in unselected, concordant populations.[13] [22]
Causal and Prospective Epidemiological Evidence
Genetic and observational studies have firmly established ApoB as a causal driver of atherosclerosis rather than a mere marker of risk.[3]
Mendelian randomization (MR): A large-scale European genome-wide association study (GWAS) using two-sample MR demonstrated that genetically determined elevations in ApoB are causally associated with coronary heart disease (CHD) (OR 1.71, 95% CI 1.53–1.91; P = 0.010), large-artery atherosclerotic stroke (ISL) (OR 1.43, 95% CI 1.23–1.66; P = 2.7×10⁻⁶), and small-vessel stroke (ISS) (OR 1.22, 95% CI 1.06–1.41; P = 0.005).[1] Multivariable MR further indicates that the clinical benefit of lipid lowering is fundamentally proportional to the absolute reduction achieved in ApoB-containing particle number, rather than the mass of LDL-C removed.[15] [16]
Prospective cohorts: In prospective analyses including the UK Biobank and secondary-prevention populations, ApoB emerged as the lipid parameter most consistently associated with myocardial infarction (MI) after adjustment for particle concentration, type, and content.[2] In primary-prevention populations (N = 389,529; median 11.1 years), each 1-SD higher ApoB concentration was associated with an adjusted hazard ratio of 1.27 (95% CI 1.15–1.40; P < .001) for incident MI, and ApoB was the only lipid measure that remained significant after full adjustment.[2]
Discordance analyses: When ApoB and LDL-C or non-HDL-C levels are discordant, prospective analyses show that ApoB retains predictive value while the cholesterol measures attenuate.[3] In the UK Biobank (N = 41,099; 9,663 MACE and 1,754 incident CAD events over ~10 years), ApoB outperformed LDL particle number: at 30% discordance the hazard ratio reached 1.4 for MACE and 2.5 for CAD, whereas discordantly elevated LDL particle number did not independently predict risk.[3]
Table 1. Genetic and prospective evidence for ApoB as a causal cardiovascular risk factor.
| Outcome measure | Study type & source | Statistical estimate | Significance |
| Coronary heart disease (CHD) | European GWAS / Mendelian randomization [1] | OR 1.71 (1.53–1.91) | P = 0.010 |
| Large-artery atherosclerotic stroke (ISL) | European GWAS / Mendelian randomization [1] | OR 1.43 (1.23–1.66) | P = 2.7×10⁻⁶ |
| Small-vessel stroke (ISS) | European GWAS / Mendelian randomization [1] | OR 1.22 (1.06–1.41) | P = 0.005 |
| Myocardial infarction (MI) | Primary-prevention cohort [2] | aHR 1.27 (1.15–1.40) per 1-SD ApoB | P < .001 |
| Discordant risk (ApoB vs. LDL particle no.) | UK Biobank discordance analysis [3] | HR up to 2.5 (CAD) for ApoB at 30% discordance | P < .0001 |
2. Insulin Resistance and Prediabetes: Pathophysiology of Glycemic Dysregulation and Lipid Dysmetabolism
Insulin resistance represents a state of impaired physiological cellular responsiveness to insulin, particularly within skeletal muscle, adipose tissue, and the liver.[4] To overcome this resistance, pancreatic beta-cells upregulate insulin secretion, producing chronic compensatory hyperinsulinemia. Over time, pancreatic capacity is overwhelmed, culminating in progressive glycemic dysregulation from normal glucose tolerance to prediabetes and overt type 2 diabetes (T2DM).[10]
The Progression of Glycemic Dysregulation
Clinical cross-sectional data confirm that elevated ApoB is independently associated with worsening glycemic parameters even in non-diabetic individuals.[5] Higher ApoB correlates with elevated fasting plasma glucose (β ≈ 2.07 mg/dL per 1-SD ApoB), higher glycated hemoglobin (β ≈ 0.06%), and increased HOMA-IR (β ≈ 0.54; all P < 0.001).[5] Individuals in the highest ApoB quartile exhibit a significantly higher odds of prediabetes than those in the lowest quartile (adjusted OR 1.53, 95% CI 1.22–1.91; P < 0.001).[5]
In apparently healthy normoglycemic cohorts (N = 7,427), positive correlations exist between metabolic syndrome, insulin resistance, and atherogenic markers (ApoB, total cholesterol/HDL-C, and LDL-C/HDL-C ratios), while negative correlations are found for cardioprotective indices such as ApoA-I, ApoA-I/ApoB, and HDL-C/ApoA-I.[6] This underscores that the lipid remodeling of insulin resistance is active and prevalent before any formal diagnosis of diabetes.[6]
Alterations in Lipoprotein Metabolism
Insulin resistance reshapes systemic lipid metabolism through several pathways:
- Unrestrained lipolysis: In insulin-resistant adipose tissue, insulin fails to suppress hormone-sensitive lipase (HSL), producing an uncontrolled flux of free fatty acids (FFAs) into the portal circulation.[7]
- Hepatic overproduction of VLDL: The influx of FFAs to the liver stimulates the synthesis and secretion of large, triglyceride-rich VLDL₁ particles.[7]
- Hypertriglyceridemia: Elevated hepatic VLDL secretion combined with downregulation of insulin-stimulated lipoprotein lipase (LPL) impairs clearance of triglyceride-rich lipoproteins (TRLs), producing persistent hypertriglyceridemia.[7]
- Remodeling to small, dense LDL: Under hypertriglyceridemic conditions, cholesteryl ester transfer protein (CETP) transfers triglycerides from VLDL to LDL and HDL in exchange for cholesteryl esters. Subsequent hepatic-lipase hydrolysis of these triglyceride-enriched particles yields small, dense LDL (sdLDL) and unstable HDL that is rapidly cleared by the kidney, depressing HDL-C.[9]
- ApoB particle amplification: Because hepatic VLDL secretion rises and remnant clearance is impaired, the absolute number of circulating ApoB-containing particles increases substantially, even if LDL-C mass remains static or declines.[7]
Systemic Pathophysiological Mechanisms
Insulin resistance accelerates vascular pathology through several non-lipid pathways:
- Endothelial dysfunction: Impaired insulin-receptor signaling in endothelial cells downregulates endothelial nitric oxide synthase (eNOS), reducing nitric oxide bioavailability, impairing vasodilation, and promoting a pro-coagulant, adhesive endothelial phenotype.[8]
- Oxidative stress and chronic inflammation: Hyperglycemia and lipid excess drive mitochondrial overproduction of reactive oxygen species (ROS), activating pro-inflammatory transcription factors and elevating systemic markers such as high-sensitivity C-reactive protein (hsCRP) and interleukin-6 (IL-6).[10]
- Plaque instability: Chronic vascular inflammation promotes macrophage infiltration and foam-cell formation; these cells secrete matrix metalloproteinases (MMPs) that degrade the collagenous fibrous cap, predisposing lesions to rupture and thrombosis.[8]
3. Why Elevated ApoB Is Particularly Dangerous in Insulin-Resistant Individuals
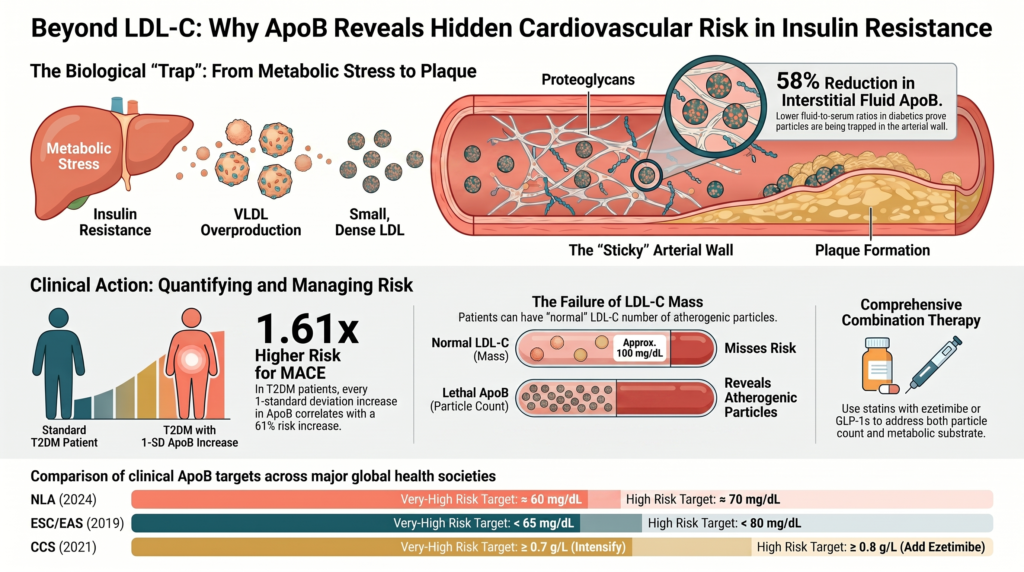
The coexistence of insulin resistance and elevated ApoB creates a highly destructive vascular environment. Insulin resistance does not merely increase the concentration of atherogenic ApoB particles; it alters both the physical properties of the particles and the structural biology of the arterial wall, multiplying the risk of particle entrapment and subsequent plaque development.[7]
Arterial Proteoglycan Remodeling
In insulin-resistant states, the subendothelial extracellular matrix of the arterial wall undergoes profound remodeling. Chronically elevated insulin and inflammatory cytokines stimulate vascular smooth-muscle cells to overproduce specific proteoglycans, notably biglycan and chondroitin-sulfate proteoglycans.[7] Insulin resistance also alters the enzymatic processing of these proteoglycans, producing longer glycosaminoglycan (GAG) chains and increased GAG sulfation.[7]
Because GAGs are highly negatively charged, this structural remodeling dramatically increases the “stickiness” of the subendothelial space, creating a physical trap that captures circulating atherogenic lipoproteins — a direct extension of the response-to-retention model of atherogenesis.[7] [17]
Enhanced Binding Affinity of Small, Dense Lipoproteins
Simultaneously, the physical properties of ApoB-containing lipoproteins are modified. The sdLDL particles and VLDL remnants predominant in insulin resistance expose positively charged amino-acid segments on the ApoB-100 protein shell.[18] These positive domains form strong electrostatic complexes with the negatively charged sulfate groups on the elongated GAG chains of arterial proteoglycans.[7]
Consequently, sdLDL and remnant particles exhibit significantly higher binding affinity for the vascular wall than larger, buoyant LDL particles.[7]
Interstitial Evidence of Transvascular Entrapment
To test this entrapment model in humans, researchers have measured lipoprotein concentrations in peripheral interstitial fluid (IF) relative to serum.[19] Because interstitial fluid drains the vascular wall, particles that traverse the endothelium without being trapped should appear in the IF.[19]
Studies show that the interstitial-fluid-to-serum ratio of ApoB is approximately 58% lower in patients with type 2 diabetes than in healthy controls (≈0.14 vs. ≈0.33).[19] This discrepancy provides direct physical evidence of enhanced transvascular retention and entrapment of ApoB-containing lipoproteins within the subendothelial matrix of insulin-resistant individuals.[19] Skin-biopsy studies further reveal higher unesterified cholesterol in diabetic subjects than in controls, corroborating tissue accumulation of cholesterol.[19]
Prolonged subendothelial entrapment amplifies the atherogenic cascade: trapped particles are exposed to local oxidative enzymes, secretory sphingomyelinase (SMase), and matrix proteases that modify them into oxidized LDL, inducing macrophage recruitment and foam-cell formation and driving rapid, silent atherosclerosis.[19] This explains why advanced, vulnerable plaques can accumulate rapidly in insulin-resistant patients even before they progress to clinical diabetes.[7]
Synergistic Interaction and Residual Cardiovascular Risk
When insulin resistance and elevated ApoB occur together, mechanistic and preclinical evidence suggests their combined impact on atherogenesis may be amplifying rather than merely additive.[7] Under normal metabolic conditions, a high concentration of ApoB-containing particles may circulate with a lower probability of arterial retention. In the presence of insulin resistance, however, the altered proteoglycan matrix and the prevalence of highly adhesive sdLDL mean that even moderate concentrations of circulating ApoB are rapidly captured, driving silent atherosclerosis.[7]
The Mismatch of Normal LDL-C and Elevated ApoB
This interaction is most problematic when patients present with normal or optimal LDL-C mass but elevated ApoB and underlying insulin resistance.[9] A standard lipid panel suggests low risk, while the elevated ApoB particle count combined with metabolic dysfunction drives progressive vascular damage.[1] The ApoB/LDL-C ratio serves as a reliable surrogate for this small-dense-LDL phenotype and is elevated in type 2 diabetes (≈0.81 ± 0.18 vs. ≈0.74 ± 0.15 in non-diabetics; P < 0.001).[9]
This synergy contributes heavily to “residual cardiovascular risk” — the persistent event rate in patients with diabetes, metabolic syndrome, or insulin resistance who have reached target LDL-C on standard statin therapy.[11] Statins upregulate LDL receptors and clear large, cholesterol-rich LDL particles but often leave a high concentration of small, dense LDL, VLDL remnants, and Lp(a) in circulation.[20]
Residual risk is, however, multifactorial, and ApoB-containing particle burden is only one of its drivers. Lipoprotein(a), systemic inflammation (reflected in elevated hsCRP), hypertension, chronic kidney disease, visceral adiposity, and lifestyle factors all contribute independently and frequently coexist with the insulin-resistant lipoprotein phenotype.[11] A comprehensive risk-reduction strategy therefore addresses these contributors alongside ApoB lowering rather than treating particle burden in isolation.[26]
Formulating Risk-Weighted ApoB
To address this limitation, researchers formulated the “risk-weighted ApoB” (RW-apoB) metric, which weights ApoB-containing subfractions (triglyceride-rich lipoprotein remnants, Lp(a), and LDL ApoB) by their relative atherogenicity to capture the elevated hazard of remnant and modified particles in a single value.[20]
In a UK Biobank derivation cohort not on lipid-lowering therapy (N = 285,060), RW-apoB reclassified roughly one-third of individuals in its top quintile relative to measured ApoB alone, identifying patients misclassified as lower-risk who nonetheless experienced a high CHD event rate of about 5.4% (vs. 3.9% in the top measured-ApoB quintile).[20] RW-apoB consistently outperformed ApoB in predicting CHD across statin-treated and high-risk cohorts (higher Harrell’s C-index; P < .0001), offering a practical tool to identify residual risk.[20]
Table 2. ApoB versus risk-weighted ApoB (RW-apoB) for residual-risk stratification.
| Stratification metric | Clinical purpose | Reclassification | CHD event rate (top quintile) |
| ApoB concentration [20] | Counts all atherogenic particles | Reference standard | ~3.9% (overlooks high-risk remnants) |
| Risk-weighted ApoB (RW-apoB) [20] | Weights ApoB, remnants, and Lp(a) by atherogenicity | Reclassified ~1/3 of top-quintile subjects | ~5.4% (captures high remnant risk) |
4. Quantitative Analysis of Major Adverse Cardiovascular Events (MACE)
Large-scale cohort studies and clinical trials have quantified the individual and joint predictive value of ApoB and insulin-resistance markers for major adverse cardiovascular events (MACE), myocardial infarction, stroke, and mortality.[1]
Evidence from Large-Scale Prospective Cohorts
In a prospective cohort of 11,918 UK Biobank participants with type 2 diabetes and no baseline ASCVD followed for a median of 185 months (~15.4 years), both ApoB and “excess ApoB” (observed ApoB minus the ApoB predicted from LDL-C in a statin-naïve reference population) were associated with incident ASCVD and MACE — linearly for ApoB and with a J-shaped relationship for excess ApoB.[21]
Compared with the lowest 50th-percentile reference, participants with higher ApoB and excess ApoB showed marked risk increases (per 1-SD ApoB: HR 1.61, 95% CI 1.40–1.85; per 1-SD excess ApoB: HR 1.18, 95% CI 1.13–1.23), particularly among statin-treated patients and those with low LDL-C.[21]
In a high-risk cohort of coronary artery disease (CAD) patients followed for roughly a decade, the baseline ApoB/LDL-C ratio (reflecting small LDL particle size and metabolic dysfunction) significantly predicted future cardiovascular events independently of type 2 diabetes status.[9]
In a multivariable-adjusted Cox model, both the ApoB/LDL-C ratio and T2DM were strong, independent predictors of events, with standardized adjusted hazard ratios of 1.17 (95% CI 1.05–1.30; P = 0.005) and 1.49 (95% CI 1.26–1.75; P < 0.001), respectively.[12]
Predictive Power of Glycemic and Lipoprotein Markers
Alternative clinical markers of insulin resistance — the triglyceride-glucose body-mass index (TyG-BMI), the triglyceride-glucose index (TyG), and the TG/HDL-C ratio — show strong prognostic value for MACE.[10] In a prospective cohort of 1,688 premature-MI (PMI) patients followed for a median of 17.4 months, those in the highest TyG-BMI quartile carried nearly threefold the MACE risk of the lowest quartile (HR 2.88, 95% CI 1.83–4.53).[10] The association was amplified in patients with comorbid diabetes (HR 3.85, 95% CI 1.79–8.27) and those with systemic inflammation (high hsCRP) (HR 3.38, 95% CI 1.78–6.43); the TyG index was also predictive (HR 1.77, 95% CI 1.11–2.82), while TG/HDL-C did not reach significance (HR 1.44, 95% CI 0.93–2.22).[10]
The Relationship Between Low LDL-C/ApoB and All-Cause Mortality
A frequently cited epidemiological observation is the U-shaped or inverse association between very low LDL-C and mortality reported in some general-population and elderly cohorts, where low LDL-C tracks with frailty, malnutrition, and chronic illness.[15] In a US NHANES analysis (2005–2016; N = 15,380; median follow-up 101 months; 1,771 deaths), both low ApoB and low LDL-C were associated with higher all-cause and cardiovascular mortality: relative to ApoB < 90 mg/dL, an ApoB ≥ 90 mg/dL carried a hazard ratio of 0.79 (95% CI 0.69–0.89) for all-cause mortality.[15]
Interpretive caution. This inverse signal is widely attributed to reverse causation and confounding by frailty and subclinical disease in unselected populations, rather than a protective effect of high atherogenic-particle burden. It does not contradict the causal, dose-dependent relationship between ApoB and atherosclerosis established by Mendelian randomization and randomized lipid-lowering trials.[1] [16] In statin-treated and secondary-prevention populations, ApoB remains a direct, positive indicator of residual cardiovascular and mortality risk, which is precisely why lowering ApoB — not merely LDL-C mass — is the therapeutic objective.[22]
Table 3. Risk estimates for ApoB and insulin-resistance markers across clinical cohorts.
| Clinical cohort | Analyzed biomarkers | Adjusted risk estimate (95% CI) | Endpoint |
| UK Biobank, T2DM [21] | ApoB vs. excess ApoB (per 1-SD) | ApoB HR 1.61 (1.40–1.85); excess ApoB HR 1.18 (1.13–1.23) | Incident ASCVD (2,548) & MACE (1,205) |
| High-risk CAD [12] | ApoB/LDL-C ratio & diabetes status | ApoB/LDL-C HR 1.17 (1.05–1.30, P=0.005); T2DM HR 1.49 (1.26–1.75, P<0.001) | CV events over ~10 years |
| Premature MI [10] | TyG-BMI, TyG, TG/HDL-C (Q4 vs Q1) | TyG-BMI HR 2.88 (1.83–4.53); TyG 1.77 (1.11–2.82); TG/HDL-C 1.44 (0.93–2.22, NS) | Incident MACE over 17.4 mo |
| US NHANES [15] | Serum ApoB / LDL-C | ApoB ≥90 vs <90 mg/dL: HR 0.79 (0.69–0.89) — low ApoB tracks higher mortality (reverse causation) | All-cause mortality (~8.4 yr) |
5. Guidelines and Risk Stratification: Integrating ApoB and Insulin Resistance
Recognizing the superior predictive value of ApoB over conventional lipids, major cardiovascular and lipid societies have integrated ApoB measurement and target values into their risk-stratification guidelines, particularly for patients with metabolic dysfunction.[11]
Comparison of Consensus Guidelines and Targets
Professional societies offer varying recommendations based on patient risk profile:
National Lipid Association (NLA): The 2024 NLA expert consensus on ApoB recommends treatment thresholds of ≈60 mg/dL (very-high risk), ≈70 mg/dL (high risk), and ≈90 mg/dL (borderline-to-intermediate risk), and classifies ApoB ≥130 mg/dL (≈90th percentile) as a risk-enhancing factor. ApoB is recommended for initial CVD-risk assessment or as an optional secondary target.[23]
European Society of Cardiology / European Atherosclerosis Society (ESC/EAS): The 2019 ESC/EAS guideline sets secondary ApoB goals of <65 mg/dL (very-high risk), <80 mg/dL (high risk), and <100 mg/dL (moderate risk), corresponding to LDL-C goals of <55, <70, and <100 mg/dL. It endorses ApoB as an alternative primary screening measurement, especially in patients with metabolic syndrome, type 2 diabetes, obesity, hypertriglyceridemia, or very low LDL-C.[24]
Canadian Cardiovascular Society (CCS): The 2021 CCS guideline prefers ApoB or non-HDL-C over LDL-C when triglycerides exceed 1.5 mmol/L (~133 mg/dL), in both primary and secondary prevention, with intensification thresholds of ApoB ≥0.8 g/L (add ezetimibe) and ≥0.7 g/L (further intensification).[25]
American College of Cardiology / American Heart Association (ACC/AHA): The 2026 ACC/AHA multisociety dyslipidemia guideline adopts the PREVENT-ASCVD equations to estimate 10- and 30-year risk in adults 30–79 years without ASCVD. LDL-C goals are <100 mg/dL (borderline/intermediate), <70 mg/dL (high risk), and <55 mg/dL (very-high-risk secondary prevention). Measuring ApoB is reasonable (Class 2a) in adults on lipid-lowering therapy — particularly those with ASCVD, cardiovascular-kidney-metabolic (CKM) syndrome, T2DM, or high triglycerides — to guide intensification once LDL-C and non-HDL-C goals are met.[26]
Table 4. Comparison of society ApoB targets and recommended application.
| Society | Very-high risk / secondary prevention | High risk / primary prevention | Recommended application |
| NLA [23] | ApoB ~60 mg/dL | ApoB ~70 mg/dL (90 mg/dL borderline-intermediate) | Optional secondary target; ≥130 mg/dL is a risk-enhancing factor. |
| ESC/EAS [24] | ApoB <65 mg/dL | ApoB <80 mg/dL (<100 moderate) | Alternative primary screening/diagnostic target; preferred in metabolic syndrome, T2DM, obesity. |
| CCS [25] | ApoB ≥0.7 g/L (intensify) | ApoB ≥0.8 g/L (add ezetimibe) | Preferred over LDL-C when TG >1.5 mmol/L (~133 mg/dL). |
| ACC/AHA [26] | Aligned with LDL-C <55 mg/dL | LDL-C <70 mg/dL (high); <100 mg/dL (intermediate) | Class 2a; guides intensification after LDL-C goals in CKM syndrome, diabetes, high TG. |
Routine Measurement Criteria in High-Risk Populations
There is a strong clinical rationale for measuring ApoB routinely in patients with insulin resistance, obesity, prediabetes, or metabolic syndrome.[11] In these populations, hepatic overproduction of VLDL and lipid remodeling produce a high concentration of small, dense LDL particles.[7]
Because these particles carry less cholesterol per particle, standard LDL-C measurements can appear normal or low, masking a high concentration of atherogenic particles.[1] Measuring ApoB directly quantifies this particle burden, identifying high-risk individuals who would otherwise be misclassified as low-risk by traditional lipid panels.[1]
This utility is recognized in the ACC/AHA dyslipidemia guideline, which incorporates the PREVENT-ASCVD equations to estimate 10- and 30-year cardiovascular risk and recommends ApoB measurement (Class 2a) to evaluate residual risk and guide intensification in patients with CKM syndrome, type 2 diabetes, or elevated triglycerides who have already reached their LDL-C and non-HDL-C goals.[26]
6. Clinical Implications and Therapeutic Pathways
In an insulin-resistant patient, elevated ApoB may signal higher residual atherogenic risk than LDL-C alone conveys; however, treatment intensity should be guided by overall ASCVD risk and guideline-defined indications rather than assuming equivalence to established type 2 diabetes.[11] The underlying pathophysiology — vascular-matrix remodeling, endothelial dysfunction, and accelerated subendothelial retention of ApoB-containing particles — is mechanistically active during the insulin-resistant and prediabetic phases and is supported by mechanistic and preclinical work, which can drive subclinical atherosclerosis before diabetes is clinically diagnosed.[7]
Statins and the Challenge of Residual Risk
Statins inhibit hepatic HMG-CoA reductase, reducing intracellular cholesterol synthesis and upregulating hepatic LDL receptors, which enhances clearance of circulating ApoB-containing particles.[27] High-intensity statins are recommended to achieve substantial LDL-C reduction.[26] However, in insulin-resistant patients, statins can leave a high concentration of residual small, dense LDL and remnant particles, underscoring the need for combination therapy.[11]
Targeted Non-Statin Lipid-Lowering Therapies
Targeted non-statin therapies further reduce ApoB and lower cardiovascular events:
- Ezetimibe: By inhibiting the NPC1L1 transporter in the small intestine, ezetimibe reduces cholesterol absorption.[27] In insulin-resistant animal models, ezetimibe — alone or with simvastatin — reduced ex vivo arterial retention of intestinal-derived ApoB-48 and ApoB-100 remnant lipoproteins, limiting cholesterol deposition.[28]
- PCSK9 inhibitors: This class includes the monoclonal antibodies alirocumab and evolocumab (and the emerging agent tafolecimab) and the small interfering RNA inclisiran.[29] By preventing PCSK9-mediated degradation of LDL receptors, these agents preserve receptor recycling, lowering LDL-C by roughly 50–60% with corresponding reductions in ApoB.[29] The oral PCSK9 inhibitor enlicitide decanoate (20 mg once daily) reduced LDL-C by ~55.8% versus placebo at 24 weeks in the phase-3 CORALreef Lipids trial, with an accompanying reduction in ApoB.[30]
- ANGPTL3 inhibitors: Angiopoietin-like protein 3 (ANGPTL3) regulates plasma lipid metabolism by inhibiting lipoprotein lipase and endothelial lipase.[31] Evinacumab, a monoclonal antibody against ANGPTL3 (a distinct target from PCSK9), lowers LDL-C by approximately 47–49% in patients with refractory homozygous familial hypercholesterolemia (HoFH).[31] Emerging antisense oligonucleotides (vupanorsen) and siRNA therapies (zodasiran, solbinsiran) target triglyceride metabolism, achieving triglyceride reductions exceeding 50% alongside modest LDL-C reductions (≤16%).[32] In TRANSLATE-TIMI 70, vupanorsen produced no clear dose-dependent LDL-C/ApoB benefit and was associated with dose-dependent increases in hepatic fat content (relative increase up to ~76%), halting its development.[32]
- Bempedoic acid: An oral prodrug activated specifically in the liver by ACSVL1 — an enzyme absent in skeletal muscle, avoiding statin-associated myopathy — that inhibits ATP-citrate lyase (ACLY) upstream of HMG-CoA reductase.[27]
In the CLEAR Outcomes trial (13,970 statin-intolerant patients at or at high risk for ASCVD), bempedoic acid (180 mg once daily) lowered LDL-C by ~21.1% and hsCRP by ~22.2% at 6 months.[33] Over a median 40.6 months, it reduced the primary 4-component MACE endpoint (CV death, non-fatal MI, non-fatal stroke, or coronary revascularization) by 13% (HR 0.87, 95% CI 0.79–0.96; P = 0.004).[33] It also reduced 3-component MACE (HR 0.85, 0.76–0.96), MI (HR 0.77, 0.66–0.91), and coronary revascularization (HR 0.81, 0.72–0.92); in the primary-prevention subgroup the 4-component MACE HR was 0.70 (95% CI 0.55–0.89).[33]
Incretin-Based Metabolic Modification
Incretin-based therapies such as semaglutide and tirzepatide achieve weight loss and improve cardiovascular risk profiles.[34]
- Semaglutide: In the SELECT trial (N = 17,604 overweight or obese patients with pre-existing CVD but without diabetes), semaglutide 2.4 mg weekly reduced 3-point MACE by 20% (HR 0.80, 95% CI 0.72–0.90; P < 0.001) over a mean 39.8 months. Reductions in inflammatory and atherogenic markers (including hsCRP and ApoB) have been reported in SELECT analyses but were secondary to the primary cardiovascular-outcome benefit and should be cited to those specific analyses.[34]
- Tirzepatide: In the SURMOUNT-1 program, tirzepatide produced weight loss up to ~20.9% and improved insulin sensitivity.[35] In a phase-2b lipoprotein analysis in type 2 diabetes, tirzepatide reduced large triglyceride-rich lipoprotein particles, small LDL particles, and the lipoprotein insulin-resistance (LPIR) score, with ApoB reductions up to ~17% and triglyceride reductions of ~31–35%, consistent with systemic metabolic and particle-level correction.[36]
- SGLT2 inhibitors: By promoting glucosuria and osmotic diuresis, SGLT2 inhibitors improve glycemic control, lower body weight and fat, and reduce cardiovascular and renal mortality; they also shift substrate utilization from carbohydrate toward lipid and ketone-body oxidation.[37]
- Dietary and lifestyle interventions: Registered dietitian nutritionist (RDN) referral is recommended for patients with elevated triglycerides (fasting TG ≥1000 mg/dL, COR 1; 150–999 mg/dL with CKM syndrome, COR 2a) to provide evidence-based counseling and reduce pancreatitis risk.[26]
Table 5. Summary of lipid-lowering and metabolic therapies relevant to ApoB reduction.
| Agent | Efficacy on ApoB / lipids | Trial | Outcome result | Safety / tolerability |
| Bempedoic acid [33] | LDL-C −21.1%, hsCRP −22.2% | CLEAR Outcomes (13,970 statin-intolerant) | MACE-4 HR 0.87 (0.79–0.96); primary prevention 0.70 (0.55–0.89) | Muscle symptoms ≈ placebo; mild ↑ uric acid, gout, cholelithiasis |
| Semaglutide [34] | MACE reduction; biomarker effects not primary endpoint | SELECT (17,604 obese CVD, no diabetes) | 3-point MACE HR 0.80 (0.72–0.90) | Well tolerated; GI effects common; sustained weight loss |
| Tirzepatide [36] | ApoB up to ~17%↓; TG ~31–35%↓; ↓LPIR | SURMOUNT / SURPASS / phase-2b | ↓ LPIR score, small LDLP, large TRLP | GI side effects; weight loss up to ~20.9% |
| Evinacumab (anti-ANGPTL3) [31] | LDL-C ≈47–49%↓ in HoFH | ELIPSE HoFH | Marked LDL-C reduction in refractory HoFH | Well tolerated; IV administration |
| Vupanorsen (ANGPTL3 ASO) [32] | TG ≈41–57%↓; LDL-C ≤16%↓ | TRANSLATE-TIMI 70 | No clear LDL-C/ApoB dose-response; development halted | Dose-dependent ↑ hepatic fat (relative up to ~76%) |
7. Discussion and Conclusion
This analysis demonstrates that evaluating cardiovascular danger requires looking beyond simple lipid-concentration mass.[4] When insulin resistance and elevated ApoB coexist, they drive silent, progressive atherogenesis through several interconnected metabolic and vascular pathways.[7]
Figure 1. Integrated pathway from chronic metabolic stress to acute cardiovascular events.
| CHRONIC METABOLIC STRESS (Insulin Resistance) |
▼
| Lipid Deregulatory Pathway
(Hepatic VLDL Excess) |
Vascular Remodeling Pathway
(Endothelial Injury) |
▼
| Elevated ApoB Count
(sdLDL & Remnants) |
Intimal Proteoglycan
Remodeling (Sticky GAGs) |
▼
| ACCELERATED TRANSVASCULAR RETENTION & SUBENDOTHELIAL TRAPPING
(~58% lower IF/serum ApoB ratio in T2DM) |
▼
| OXIDATIVE MODIFICATION & MONOCYTE RECRUITMENT |
▼
| FOAM-CELL FORMATION & PLAQUE PROGRESSION |
▼
| PLAQUE RUPTURE & THROMBOSIS |
▼
| ACUTE CARDIOVASCULAR EVENTS (MACE, MI, Stroke) |
Discussion: a unified model and its limits
The evidence reviewed here converges on a single coherent model, summarized in Figure 1. Chronic insulin resistance drives hepatic overproduction of triglyceride-rich VLDL and remodeling into small, dense LDL, raising the circulating ApoB particle count even when LDL-C mass appears normal; in parallel, the insulin-resistant arterial wall is remodeled into a more adhesive, proteoglycan-rich surface that preferentially retains those particles. The convergence of more atherogenic particles and a more retentive vessel wall offers a unifying explanation for why cardiovascular risk in insulin-resistant and prediabetic individuals can outpace what a standard lipid panel predicts.
Two interpretive cautions follow directly from the strength of the underlying evidence. First, the human data are strongest for the associations — the Mendelian-randomization causality of ApoB, the prospective hazard ratios, and the interstitial-fluid retention signal — while the step-by-step proteoglycan-remodeling and foam-cell sequence rests substantially on mechanistic and preclinical work, including an animal model. These mechanistic steps are biologically plausible and supported, but they are not proven to the same degree in humans and are presented here as a plausible mechanism rather than established clinical fact. Second, ApoB is one driver of residual risk among several; lipoprotein(a), inflammation, hypertension, chronic kidney disease, adiposity, and lifestyle act independently and should be managed alongside it.
Conclusion and clinical translation
For the insulin-resistant patient, the practical message is that LDL-C alone can understate atherogenic burden, and that ApoB (or, where ApoB is unavailable, non-HDL-C) more directly captures the particle count that the biology implicates. Translating this into care does not require treating prediabetes as though it were established diabetes; it requires measuring the right thing and acting on overall risk.
Four principles summarize the clinical translation:
- Measure particle burden. Consider ApoB (or non-HDL-C) in patients with insulin resistance, obesity, prediabetes, or metabolic syndrome, in whom LDL-C most often underestimates atherogenic-particle number.[11]
- Match intensity to overall risk. Use elevated ApoB to refine — not replace — guideline-based risk assessment, intensifying lipid-lowering according to overall ASCVD risk and society-defined indications rather than assuming diabetes-equivalent risk.[11] [26]
- Lower ApoB with combination therapy where indicated. High-intensity statins paired with ezetimibe, PCSK9 inhibitors, or bempedoic acid maximize ApoB-containing particle clearance and reduce events in appropriate populations.[27] [33]
- Address the metabolic substrate and co-drivers. GLP-1 receptor agonists such as semaglutide reduce cardiovascular events in obesity with established CVD,[34] and the dual GIP/GLP-1 agonist tirzepatide improves insulin sensitivity and insulin-resistance lipoprotein markers;[36] these, together with control of Lp(a)-related risk, inflammation, blood pressure, and lifestyle, complete a risk-reduction strategy that treats particle burden as one component rather than the whole.
In sum, the central thesis holds: in insulin-resistant states, ApoB-defined particle burden and the biology of arterial retention together capture risk that LDL-C can miss. The appropriate response is better measurement and risk-proportionate, multifactorial treatment — stated with confidence where randomized and genetic evidence supports it, and with appropriate caution where the mechanism remains inferential.
References
Citations follow IEEE numbering. This reference list prioritizes primary peer-reviewed studies, official guideline documents, and indexed trial reports, and review or consensus documents where a full primary report was not the most appropriate source; these are identified as such.
- Kong X, Cai Y, Li Y, Wang P. Causal relationship between apolipoprotein B and risk of atherosclerotic cardiovascular disease: a mendelian randomization analysis. Health Inf Sci Syst. 2025;13(1):13. Published 2025 Jan 3. doi:10.1007/s13755-024-00323-5
- Marston NA, Giugliano RP, Melloni GEM, et al. Association of Apolipoprotein B-Containing Lipoproteins and Risk of Myocardial Infarction in Individuals With and Without Atherosclerosis: Distinguishing Between Particle Concentration, Type, and Content. JAMA Cardiol. 2022;7(3):250-256. doi:10.1001/jamacardio.2021.5083
- Epstein E, Ekpo E, Evans D, et al. Apolipoprotein B outperforms low density lipoprotein particle number as a marker of cardiovascular risk in the UK Biobank. Eur J Prev Cardiol. Published online September 1, 2025. doi:10.1093/eurjpc/zwaf554
- Borén J, Chapman MJ, Krauss RM, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2020;41(24):2313-2330. doi:10.1093/eurheartj/ehz962
- Bekbossynova M, Ivanova-Razumova T, Kali A, et al. Apolipoprotein B and Glycemic Dysregulation: New Predictors of Type 2 Diabetes in High-Cardiovascular-Risk Populations. J Pers Med. 2025;15(5):163. Published 2025 Apr 23. doi:10.3390/jpm15050163
- Sung KC, Hwang ST. Association between insulin resistance and apolipoprotein B in normoglycemic Koreans. Atherosclerosis. 2005;180(1):161-169. doi:10.1016/j.atherosclerosis.2004.11.009
- Mangat R, Warnakula S, Borthwick F, et al. Arterial retention of remnant lipoproteins ex vivo is increased in insulin resistance because of increased arterial biglycan and production of cholesterol-rich atherogenic particles that can be improved by ezetimibe in the JCR:LA-cp rat. J Am Heart Assoc. 2012;1(5):e003434. doi:10.1161/JAHA.112.003434
- Borén J, Williams KJ. The central role of arterial retention of cholesterol-rich apolipoprotein-B-containing lipoproteins in the pathogenesis of atherosclerosis: a triumph of simplicity. Curr Opin Lipidol. 2016;27(5):473-483. doi:10.1097/MOL.0000000000000330
- BARBARA LARCHER, THOMAS PLATTNER, ARTHUR MADER, ALEXANDER VONBANK, ANDREAS LEIHERER, AXEL MUENDLEIN, HEINZ DREXEL, CHRISTOPH H. SAELY; 456-P: The ApoB/LDL-C Ratio Predicts Cardiovascular Events in Coronary Artery Disease Patients Independent of Type 2 Diabetes Status. Diabetes 14 June 2024; 73 (Supplement_1): 456–P. https://doi.org/10.2337/db24-456-P
- Zhou Y, Wang Y, Chen J, Jiang L, Chu R, Tian W, Wang J, Liu Y and Gao J (2026) The association between insulin resistance indices and the occurrence of major adverse cardiovascular events in patients with premature myocardial infarction: a prospective cohort study. Front. Nutr. 13:1724362. doi: 10.3389/fnut.2026.1724362
- Soffer DE, Marston NA, Maki KC, et al. Role of apolipoprotein B in the clinical management of cardiovascular risk in adults: An Expert Clinical Consensus from the National Lipid Association. J Clin Lipidol. 2024;18(5):e647-e663. doi:10.1016/j.jacl.2024.08.013
- BARBARA LARCHER, MAXIMILIAN MAECHLER, LUKAS SPRENGER, ARTHUR MADER, ALEXANDER VONBANK, THOMAS PLATTNER, ANDREAS LEIHERER, AXEL MUENDLEIN, HEINZ DREXEL, CHRISTOPH H. SAELY; 260-OR: The ApoB/LDL-C Ratio Predicts Major Cardiovascular Events in Cardiovascular Disease Patients Independent of Type 2 Diabetes Status. Diabetes 20 June 2023; 72 (Supplement_1): 260–OR. https://doi.org/10.2337/db23-260-OR
- Sniderman AD, Thanassoulis G, Glavinovic T, et al. Apolipoprotein B Particles and Cardiovascular Disease: A Narrative Review. JAMA Cardiol. 2019;4(12):1287-1295. doi:10.1001/jamacardio.2019.3780
- Ference BA, Ginsberg HN, Graham I, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2017;38(32):2459-2472. doi:10.1093/eurheartj/ehx144
- Yu X, Yuan Y, Dong X, et al. Low apolipoprotein B and LDL-cholesterol are associated with the risk of cardiovascular and all-cause mortality: a prospective cohort. Ann Med. 2025;57(1):2529565. doi:10.1080/07853890.2025.2529565
- Richardson TG, Sanderson E, Palmer TM, et al. Evaluating the relationship between circulating lipoprotein lipids and apolipoproteins with risk of coronary heart disease: A multivariable Mendelian randomisation analysis. PLoS Med. 2020;17(3):e1003062. Published 2020 Mar 23. doi:10.1371/journal.pmed.1003062
- Williams KJ, Tabas I. The response-to-retention hypothesis of early atherogenesis. Arterioscler Thromb Vasc Biol. 1995;15(5):551-561. doi:10.1161/01.atv.15.5.551
- Camejo G, Hurt-Camejo E, Wiklund O, Bondjers G. Association of apo B lipoproteins with arterial proteoglycans: pathological significance and molecular basis. Atherosclerosis. 1998;139(2):205-222. doi:10.1016/s0021-9150(98)00107-5
- Björklund P, Härdfeldt J, Äikäs L, et al. Increased transvascular retention of atherogenic lipoproteins in type 2 diabetes relates to their enhanced proteoglycan binding. JCI Insight. 2026;11(10):e177849. Published 2026 Apr 7. doi:10.1172/jci.insight.177849
- Rehman MB, Björnson E, Adiels M, et al. Risk-weighted apoB: a novel summary metric outperforming traditional lipid biomarkers in predicting coronary heart disease. Eur Heart J. Published online January 22, 2026. doi:10.1093/eurheartj/ehaf1124
- Lyu L, Kao C, Su J, et al. Association of apolipoprotein B and excess apolipoprotein B with cardiovascular risk in type 2 diabetes: a prospective cohort study of the UK Biobank. Lipids Health Dis. 2026;25(1):55. Published 2026 Jan 15. doi:10.1186/s12944-025-02852-8
- Sniderman AD, Navar AM, Thanassoulis G. Apolipoprotein B vs Low-Density Lipoprotein Cholesterol and Non-High-Density Lipoprotein Cholesterol as the Primary Measure of Apolipoprotein B Lipoprotein-Related Risk: The Debate Is Over. JAMA Cardiol. 2022;7(3):257-258. doi:10.1001/jamacardio.2021.5080
- Soffer DE, Marston NA, Maki KC, et al. Role of apolipoprotein B in the clinical management of cardiovascular risk in adults: An Expert Clinical Consensus from the National Lipid Association. J Clin Lipidol. 2024;18(5):e647-e663. doi:10.1016/j.jacl.2024.08.013
- Mach F, Baigent C, Catapano AL, et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. 2020;41(1):111-188. doi:10.1093/eurheartj/ehz455
- Pearson GJ, Thanassoulis G, Anderson TJ, et al. 2021 Canadian Cardiovascular Society Guidelines for the Management of Dyslipidemia for the Prevention of Cardiovascular Disease in Adults. Can J Cardiol. 2021;37(8):1129-1150. doi:10.1016/j.cjca.2021.03.016
- Blumenthal RS, Morris PB, Gaudino M, et al. 2026 ACC/AHA/AACVPR/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Dyslipidemia: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J Am Coll Cardiol. 2026;87(19):2624-2757. doi:10.1016/j.jacc.2025.11.016
- Albosta M, Grant JK, Michos ED. Bempedoic Acid: Lipid Lowering for Cardiovascular Disease Prevention. Heart Int. 2023;17(2):27-34. Published 2023 Nov 1. doi:10.17925/HI.2023.17.2.1
- Mangat R, Warnakula S, Borthwick F, et al. Arterial retention of remnant lipoproteins ex vivo is increased in insulin resistance because of increased arterial biglycan and production of cholesterol-rich atherogenic particles that can be improved by ezetimibe in the JCR:LA-cp rat. J Am Heart Assoc. 2012;1(5):e003434. doi:10.1161/JAHA.112.003434
- Sabatine MS, Giugliano RP, Keech AC, et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N Engl J Med. 2017;376(18):1713-1722. doi:10.1056/NEJMoa1615664
- Ballantyne CM, Gellis L, Tardif JC, et al. Efficacy and Safety of Oral PCSK9 Inhibitor Enlicitide in Adults With Heterozygous Familial Hypercholesterolemia: A Randomized Clinical Trial. JAMA. 2026;335(2):129-139. doi:10.1001/jama.2025.20620
- Raal FJ, Rosenson RS, Reeskamp LF, et al. Evinacumab for Homozygous Familial Hypercholesterolemia. N Engl J Med. 2020;383(8):711-720. doi:10.1056/NEJMoa2004215
- Bergmark BA, Marston NA, Bramson CR, et al. Effect of Vupanorsen on Non-High-Density Lipoprotein Cholesterol Levels in Statin-Treated Patients With Elevated Cholesterol: TRANSLATE-TIMI 70. Circulation. 2022;145(18):1377-1386. doi:10.1161/CIRCULATIONAHA.122.059266
- Nissen SE, Lincoff AM, Brennan D, et al. Bempedoic Acid and Cardiovascular Outcomes in Statin-Intolerant Patients. N Engl J Med. 2023;388(15):1353-1364. doi:10.1056/NEJMoa2215024
- Lincoff AM, Brown-Frandsen K, Colhoun HM, et al. Semaglutide and Cardiovascular Outcomes in Obesity without Diabetes. N Engl J Med. 2023;389(24):2221-2232. doi:10.1056/NEJMoa2307563
- Jastreboff AM, Aronne LJ, Ahmad NN, et al. Tirzepatide Once Weekly for the Treatment of Obesity. N Engl J Med. 2022;387(3):205-216. doi:10.1056/NEJMoa2206038
- Wilson JM, Nikooienejad A, Robins DA, et al. The dual glucose-dependent insulinotropic peptide and glucagon-like peptide-1 receptor agonist, tirzepatide, improves lipoprotein biomarkers associated with insulin resistance and cardiovascular risk in patients with type 2 diabetes. Diabetes Obes Metab. 2020;22(12):2451-2459. doi:10.1111/dom.14174
- Szekeres Z, Toth K, Szabados E. The Effects of SGLT2 Inhibitors on Lipid Metabolism. Metabolites. 2021;11(2):87. Published 2021 Feb 1. doi:10.3390/metabo11020087