Clinical and Mechanistic Significance of Absent Coronary Plaque in a Young Adult with Extreme Diet-Induced Hypercholesterolemia: A Critical Appraisal of the Lean Mass Hyper-Responder Phenotype
Highlights
- A single plaque-free coronary CT angiogram after ~6.7 years of adult-onset extreme hypercholesterolemia does not demonstrate protection from ApoB-mediated atherosclerosis.
- Reconstructed cumulative exposure (~6,185 mg/dL-years at age 37) is well below age-matched untreated heterozygous familial hypercholesterolemia and far below homozygous FH.
- Zero detectable plaque is common in young people with lifelong severe LDL elevation and is expected during the biological latency of atherosclerosis.
- A report of 0 mm³ reflects disease below the imaging/segmentation threshold, not the biological absence of atherosclerosis.
- The supporting KETO-CTA trial was formally retracted (JACC: Advances, 11 March 2026) and should no longer be considered reliable evidence.
Abstract
Background. The lean mass hyper-responder (LMHR) phenotype—marked elevations in LDL cholesterol (LDL-C) and HDL-C with low triglycerides in lean, metabolically healthy individuals on ketogenic diets—has been advanced as a challenge to cumulative-exposure models of atherosclerosis. A 2026 case report described a man in his 30s with sustained extreme hypercholesterolemia (peak LDL-C 574 mg/dL; apolipoprotein B [ApoB] >300 mg/dL) for approximately six years and eight months who showed a coronary artery calcium score of zero and 0 mm³ of quantified coronary plaque on AI-analyzed coronary CT angiography (CCTA).
Approach. This critical appraisal independently re-verifies every material factual claim against peer-reviewed and official sources, reconstructs the patient’s cumulative atherogenic exposure, benchmarks it against familial hypercholesterolemia (FH) cohorts, and evaluates the imaging, mechanistic, and evidentiary basis for inferring reduced particle atherogenicity or long-term safety.
Findings. The absence of macroscopically detectable plaque after fewer than seven years of adult-onset exposure is consistent with the established latency of atherosclerosis and with the high prevalence of zero coronary calcium among young FH patients. Reconstructed exposure (~6,185 mg/dL-years) is well below age-matched untreated heterozygous FH. A 0 mm³ result denotes disease below the detection threshold rather than its absence. No peer-reviewed human evidence demonstrates reduced arterial retention of ketogenic LDL after accounting for ApoB concentration, and the principal supporting trial (KETO-CTA) has been formally retracted.
Conclusions. The available evidence most strongly supports the interpretation that this case reflects biological latency and lower cumulative exposure—not resistance to ApoB-mediated disease. Overinterpretation risks encouraging deferral of guideline-directed lipid-lowering therapy.
1. Introduction and the Index Case
The causal role of apolipoprotein B (ApoB)-containing lipoproteins in atherosclerotic cardiovascular disease (ASCVD) is among the most consistently replicated findings in cardiovascular medicine. The emergence of the lean mass hyper-responder (LMHR) phenotype—marked elevations in LDL cholesterol (LDL-C) and HDL-C with low triglycerides in lean, metabolically healthy individuals adopting ketogenic diets—has been presented as a challenge to traditional risk-stratification models. This appraisal examines whether a widely discussed 2026 case report supports that challenge.
The index report describes an athletic male in his 30s (“Patient N”) who initiated a ketogenic diet (approximately 80% fat, 18% protein, 2% carbohydrate) to manage biopsy-proven ulcerative colitis, entering clinical remission within roughly two weeks. His LDL-C rose from a mixed-macronutrient baseline of 95 mg/dL to a peak of 574 mg/dL, with total cholesterol reaching 705 mg/dL. Over approximately six years and eight months he maintained LDL-C broadly in the 400–600 mg/dL range, HDL-C of 124 mg/dL, and very low triglycerides; ApoB consistently exceeded 300 mg/dL and lipoprotein(a) [Lp(a)] ranged from roughly 100 to 194 nmol/L. On 3 February 2026 CCTA with expert reading and HeartFlow AI-enabled plaque analysis reported a coronary artery calcium (CAC) score of 0, CAD-RADS 0, and 0 mm³ of total quantified coronary plaque, placing him “in the lowest percentile” for plaque burden [1].
The authors argue the case highlights the limitations of extrapolating cardiovascular risk from LDL-C in isolation and raise the possibility that the diet-induced etiology may modify the intrinsic atherogenicity of ApoB-containing particles, while noting that a single case cannot redefine practice [1]. A case report is nonetheless the lowest tier of clinical evidence—a single, self-selected observation without control or randomization—and every subsequent inference must be read within that constraint.
2. ApoB Reporting Considerations
The report documents ApoB above 300 mg/dL, addressing a limitation of earlier LMHR discussions. Several gaps nonetheless constrain strong interpretation: no rigorous pre-diet baseline ApoB corresponding to the baseline LDL-C of 95 mg/dL is provided, and serial values are reported sporadically rather than as a time-weighted series; assay standardization against WHO/IFCC reference materials is not documented, which matters most at the extreme (>300 mg/dL) range; and direct LDL particle number, remnant particles, and VLDL size are not systematically reported, preventing assessment of discordance between LDL-C concentration and atherogenic particle number [1].
3. Cumulative LDL-C and ApoB Exposure
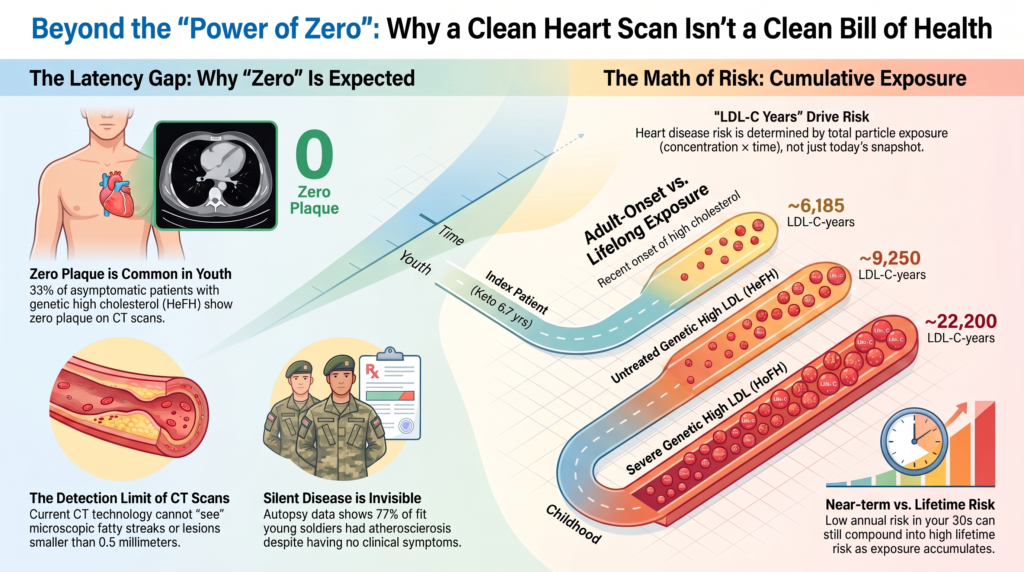
Atherosclerosis is a cumulative, dose-dependent process driven by the total number of ApoB-containing particles retained in the arterial wall over time, best quantified as “LDL-C-years” or “ApoB-years.” The log-linear, duration-dependent relationship to risk is established in consensus statements from the European Atherosclerosis Society [8,9]. For the index patient at age 37, cumulative exposure can be approximated as 95 mg/dL × 30 years + ~500 mg/dL × 6.67 years ≈ 6,185 mg/dL-years. This arithmetic is correct under the stated assumptions; the FH comparators in Table 1 are illustrative model outputs dependent on assumed lifelong LDL-C trajectories, not independently published constants.
Table 1. Illustrative cumulative LDL-C exposure (mg/dL-years) by cohort and age (middle-estimate scenarios).
| Cohort & milestone | Low | Middle | High |
| Index patient — age 30 (diet onset) | 2,850 | 2,850 | 2,850 |
| Index patient — age 37 (evaluation) | 5,518 | 6,185 | 6,851 |
| Index patient — age 50 (projected) | 10,850 | 12,850 | 14,850 |
| Untreated HeFH — age 37 | 7,030 | 9,250 | 12,950 |
| Untreated HoFH — age 37 | 14,800 | 22,200 | 29,600 |
HeFH, heterozygous familial hypercholesterolemia; HoFH, homozygous familial hypercholesterolemia. FH values are illustrative models under stated assumptions, not measured data.
Although the patient’s current LDL-C (~574 mg/dL) sits in the HoFH range, his cumulative exposure at age 37 (~6,185 mg/dL-years) is well below that of an age-matched untreated HeFH patient (~9,250) and roughly one-third that of an untreated HoFH patient (~22,200). Because atherosclerosis requires time to progress from particle retention to a macroscopically visible lesion, a clean CCTA after only ~6.7 years of adult-onset exposure is biologically consistent with existing evidence on plaque latency. Projected exposure by age 50 (~12,850 mg/dL-years) would surpass a typical HeFH patient; such projections illustrate cumulative exposure and do not predict specific events or their timing (Figure 1).
Figure 1. Cumulative LDL-C-years by age for the index case versus normolipidemic, treated/untreated heterozygous FH, and homozygous FH trajectories. The index case accrues exposure slowly until ketogenic-diet onset at age 30, then rises steeply; at evaluation (age 37) it lies below an age-matched untreated HeFH trajectory and far below untreated HoFH. FH curves are illustrative models under stated assumptions, not measured values.
4. Familial Hypercholesterolemia and the Frequency of Zero Plaque
Familial hypercholesterolemia (FH) is lifelong, congenital LDL-C elevation. Verified epidemiology: heterozygous FH (HeFH) prevalence is ~1 in 311 (95% CI 1:250–1:397) [6]; homozygous FH (HoFH) ~1 in 300,000. Without effective treatment, cumulative coronary heart disease risk by age 60 is at least ~50% in men and ~30% in women [23], and the CDC states heart attacks occur in 30% of women with FH by age 60 and 50% of men by age 50, with early treatment reducing coronary risk by ~80% [24]. The best relative-risk estimate is the Copenhagen General Population Study: ~13-fold increased CHD risk in untreated definite/probable FH (95% CI 10–17) and ~10-fold in treated FH [7]. Untreated HoFH develops ASCVD at a median age ~28 with median death ~32.
Critically, a clean scan in a young person with severe LDL elevation is the norm, not the exception. In the multinational pooled analysis by Nasir et al. of 1,011 asymptomatic HeFH patients free of clinical ASCVD, 41.8% had CAC = 0 and 33.1% had no plaque on CTA; women were more likely than men to have CAC = 0 (48.0% vs 34.6%) [4]. General-population imaging concurs: in the Multi-Ethnic Study of Atherosclerosis (MESA) analysis of participants with LDL-C ≥190 mg/dL, 37% had CAC = 0, with younger age, female sex, and absence of diabetes predicting a zero score [20]. In fairness to the opposing view, that analysis also found CAC = 0 conferred lower event risk even among high-LDL individuals; the qualification for this case is that CAC scoring detects only calcified disease and cannot exclude early non-calcified plaque, and MESA participants were, on average, decades older. The index patient’s clean scan falls squarely within the documented spectrum of subclinical-disease latency in young FH populations.
Why lifelong FH exposure is not simply “more of the same”
A fair objection is that LMHR exposure begins in adulthood whereas FH exposure is congenital—and the difference may exceed arithmetic. The developing arterial wall is not identical to the mature one: through childhood and adolescence the intima remodels and proteoglycan content changes, so exposure during these formative years may establish lipid retention and lesion architecture on a more advanced trajectory. This favors the patient in the near term while underscoring that his lower cumulative exposure, not any special particle biology, is the parsimonious explanation for the clean scan. One caveat runs the other way: FH cohorts differ from this patient in more than exposure timing—distributions of Lp(a), blood pressure, modifier genes, and diet all vary—so the comparison bounds the plausibility of his result rather than proving equivalence.
5. Latency: From Particle Exposure to Detectable Plaque
Human atherogenesis unfolds over decades, and its earliest stages are invisible to clinical imaging. Higher ApoB concentrations increase the probability of endothelial transport through concentration-dependent mechanisms that include passive flux and regulated caveolar transcytosis via receptors such as LDLR, SR-B1, and ALK1 [10]. Once in the intima, particles bind arterial proteoglycans, are chemically modified, and are taken up by macrophages that become foam cells—forming microscopic fatty streaks that cause neither calcification nor stenosis and are undetectable by CAC scanning or CCTA.
Autopsy evidence confirms this silent disease. Among U.S. soldiers killed in Korea (mean age ~22), 77.3% had gross coronary atherosclerosis despite being young and asymptomatic [11]; among Vietnam combat casualties, 45% had some coronary atherosclerosis and 5% severe disease [12]. The PDAY and Bogalusa Heart studies demonstrated that fatty-streak and raised-lesion extent in 15–39-year-olds correlated with non-HDL-C, hypertension, and obesity, and inversely with HDL-C [13,14]. A report of 0 mm³ on CCTA in a person’s 30s therefore does not indicate the biological absence of atherosclerosis; it indicates the absence of lesions advanced enough to exceed the detection limits of the technology.
6. CCTA and AI Plaque-Quantification: Detection Limits
- Spatial resolution. Clinical CT in-plane resolution is ~0.4–0.5 mm with ~0.5–0.6 mm slice thickness; newer ultra-high-resolution and photon-counting systems approach ~0.15–0.2 mm. Early atheroma (intimal thickness ~100–300 µm) lies below this floor and is lost to volume averaging.
- Quantification thresholds. SCCT standards recommend limiting quantitative plaque analysis to vessels ≥2.0 mm in diameter [22]; there is no single universally established minimum detectable plaque volume. A report of 0 mm³ does not mean zero plaque—it means plaque below the software’s segmentation threshold.
- Serial changes of a few mm³ are highly sensitive to heart rate, contrast timing, reconstruction kernel, and slice thickness; reduced epicardial fat in lean individuals lowers signal-to-noise at the vessel wall.
The AI tool used (HeartFlow) was validated in the REVEALPLAQUE study against intravascular ultrasound (IVUS) in 237 patients, with a mean IVUS total plaque volume of 186.0 mm³ and a total-plaque-volume correlation of r = 0.91 [5]. That validation was performed in an older, higher-burden, symptomatic CAD population; it establishes correlation in high-plaque arteries but does not establish that 0–5 mm³ longitudinal changes in young, low-burden arteries are biologically definitive. AI improves reproducibility and can flag plaque below confident human reading, but remains fundamentally constrained by CT spatial resolution. Plaque composition also carries prognostic weight beyond volume: in SCOT-HEART, low-attenuation plaque burden was the strongest predictor of myocardial infarction, with burden >4% associated with roughly fivefold higher risk [21]; this derives from a chest-pain referral population, so external validity to a young asymptomatic individual is limited, though the general lesson—composition matters, not only volume—holds.
7. The Retracted KETO-CTA Trial
The 2026 case report leaned in part on the KETO-CTA trial to support the claim that diet-induced ApoB elevations do not drive plaque progression. That paper received an Expression of Concern in January 2026 and was formally retracted from JACC: Advances on 11 March 2026, at the request of the authors and editors, on the grounds that methodological concerns affected the reliability of the data and the identified errors were too great to correct with a corrigendum [2]. It should no longer be considered reliable evidence.
The verified problems were substantive: the registered primary outcome (percent change in non-calcified plaque volume; NCT05733325) was not foregrounded [3]; a co-author was affiliated with the analysis vendor (Cleerly) in a role co-authors reported being unaware of, and the analysis was not double-blinded as assumed; and several authors have stated they lacked access to the raw data before publication [2]. The only peer-reviewed progression figure is a median non-calcified plaque volume increase of 18.9 mm³ (IQR 9.3–47.0) over one year, against a pre-registered conservative estimate of ~7 mm³ [3] and PARADIGM annualized percent-atheroma-volume change of ~0.45% (low-risk) and ~0.58% (high-risk) [19]. Post-retraction re-analysis figures circulating as “plaque regression” originate from non-peer-reviewed author communications and are excluded from the evidentiary basis of this appraisal.
8. Mechanistic Evaluation: The Hypothesized Reduction in Particle Atherogenicity
Stated precisely, current evidence does not support the hypothesis that ketogenic LDL particles possess reduced intrinsic atherogenicity. The mechanistic case has three parts, none of which is currently supported.
First, origin is not safety. The lipid-energy model plausibly explains why hepatic lipoprotein secretion rises under carbohydrate restriction in lean individuals, but a mechanism explaining why particles are produced says nothing about whether they are retained. Establishing benignity would require direct human kinetic evidence (e.g., stable-isotope ApoB-100 turnover) of altered residence time, which does not exist for ketogenic LMHRs.
Second, particle size does not confer immunity. Small dense (~18–20.5 nm) and large buoyant (~20.5–23 nm) LDL both traverse endothelial junctions. Small dense LDL does carry particle-level disadvantages—longer plasma residence, greater oxidation susceptibility, higher proteoglycan retention—so a shift toward larger particles is not entirely neutral; but larger particles deliver more cholesterol per particle, and the net effect does not exempt large-LDL phenotypes from atherogenicity at a given ApoB particle number. Every LDL particle carries exactly one ApoB-100 molecule, and no convincing human evidence demonstrates reduced arterial retention of ketogenic LDL after accounting for ApoB concentration.
Third, metabolic health attenuates but does not eliminate risk. Insulin sensitivity, low triglycerides, high HDL-C, and low hs-CRP alter the slope of the risk curve. Endothelial transport is shaped by shear stress, permeability, nitric-oxide signaling, inflammation, hypertension, and glycocalyx integrity, several favorable here; but the receptor-mediated pathways saturate at relatively low concentrations, so at sustained ApoB >300 mg/dL the concentration-driven component increasingly dominates [10]. Mendelian randomization consistently shows genetic ApoB/LDL elevations raise ASCVD risk log-linearly and largely independent of metabolic syndrome, diabetes, or inflammation [8,9]. At this magnitude of exposure, there is currently no evidence that favorable metabolic modifiers fully offset the retention pressure created by the concentration itself.
Two additional factors compound concern: Lp(a) of 100–194 nmol/L, each particle carrying an ApoB-100 plus pro-thrombotic, pro-calcific apo(a) and oxidized phospholipids, whose genetically determined risk metabolic health does not neutralize; and a premature paternal coronary event, compatible with inherited susceptibility beyond monogenic FH, though shared environment cannot be excluded.
9. Absolute versus Relative Risk
A large relative risk applied to a low baseline can still yield a modest short-term absolute event rate, while the same relative risk sustained over decades compounds into a large lifetime burden. At ~37, this patient’s annual absolute probability of a hard coronary event is low because baseline risk in the late 30s is low—precisely why a clean scan and absence of symptoms are unremarkable now, and why they provide little reassurance about the decades ahead. As an illustration, if extreme sustained ApoB raised the annual hard-event probability from a baseline near 0.05% to roughly 0.4–0.5%, that is a large relative increase yet still small in any single year; integrated across remaining life, the same multiplier compounds into a substantially elevated cumulative probability. (These figures are illustrative, not derived from the patient’s data.) The relevant quantity is cumulative lifetime exposure and the lifetime event curve it drives, not this year’s event probability.
10. Lipid-Lowering Therapy: The Evidence Against Deferral
The Cholesterol Treatment Trialists’ meta-analyses show a ~22% reduction in major vascular events per 1.0 mmol/L (38.7 mg/dL) LDL-C reduction [15], and ~23% for major coronary events [16], proportional to absolute lowering. Regarding safety, the CTT individual-participant meta-analysis found statins caused only a ~7% relative increase in muscle-symptom reports in year 1 (RR 1.07, 95% CI 1.04–1.10) with no excess thereafter, concluding that >90% of muscle-symptom reports were not due to the statin [17]; the National Lipid Association estimates pharmacological statin-associated muscle symptoms at ~1–2% [18]. In accordance with ACC/AHA and ESC guidance, sustained severe ApoB/LDL-C elevation of this magnitude is a strong indication for lipid-lowering therapy, independent of a single reassuring scan.
11. Structured Appraisal of the Central Questions
Is zero detectable plaque after ~6 years 8 months of extreme exposure biologically surprising in a man in his 30s?
Assessment: Not supported. A clean scan after ~6.7 years of adult-onset exposure is biologically consistent with plaque latency—an apparent delay in clinically detectable plaque formation, not a demonstration of resistance.
Is the result outside the range observed in young people with FH?
Assessment: Not supported. Multinational FH registries and general-population imaging show a large share of comparable individuals have CAC = 0 and no CCTA-detectable plaque; this case falls within that spectrum.
Does the result show LMHR ApoB particles are less atherogenic?
Assessment: Not supported. No biophysical evidence indicates reduced proteoglycan affinity or oxidation susceptibility per ApoB particle; the one longitudinal LMHR dataset, now retracted, showed substantial non-calcified plaque progression.
Does it establish that continuing the exposure is safe?
Assessment: Not supported. A single cross-sectional scan in the early latency phase cannot predict safety over subsequent decades as cumulative exposure rises.
What evidence would be needed before advising the public that extreme diet-induced ApoB may be acceptable?
Assessment: A high bar, not yet met. Prospective, well-matched, multi-year cohort evidence with high-resolution imaging and hard endpoints, demonstrating absence of progression and event risk versus normolipidemic controls.
Is the manuscript’s interpretation appropriately cautious?
Assessment: It could reasonably mislead. Framing such as “apparent resistance” and reliance on a now-retracted trial oversteps the data and could encourage deferral of guideline-directed therapy.
12. Conclusions and a Path Forward
The zero-plaque finding is best understood as a marker of early latency in a young, active individual who has not yet accumulated the cumulative atherogenic exposure required to cross CCTA’s detection threshold. Favorable metabolic markers attenuate risk; they do not repeal the mass-action mechanics of subendothelial ApoB retention. Guideline-directed lipid-lowering therapy remains indicated.
Resolving whether the LMHR phenotype carries genuine resilience will require a prospective, multicenter, registry-matched cohort comparing metabolically healthy LMHR individuals against genetically confirmed HeFH and normolipidemic controls, with CCTA at baseline, year 3, and year 5 using ultra-high-resolution or photon-counting CT; fully blinded independent core-lab analysis across multiple validated platforms; primary endpoints of absolute non-calcified plaque-volume change and percent-atheroma-volume change; and secondary endpoints including low-attenuation plaque, endothelial function, ApoB kinetics, and inflammatory biomarkers. Until such evidence exists, a single plaque-free scan should not be read as evidence of safety.
Declarations
Funding.
This work received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors. [Author to confirm or amend.]
Competing interests.
The author is the founder and operator of Curing Heart Disease, LLC, a cardiovascular health-education platform (curingheartdisease.com), which represents a potential non-financial and reputational interest in the subject matter. The author declares no financial relationships with pharmaceutical or medical-device manufacturers relevant to this work. [Author to confirm and disclose any additional interests.]
Data availability.
This is a critical appraisal of previously published literature; no new primary data were generated. All sources are cited and publicly available through the referenced journals and official records. The raw imaging artifacts underlying the index case report were not available to the author and could not be independently re-analyzed.
Ethics approval and consent.
This appraisal analyzes an already-published, de-identified case report and other published literature; it did not involve new human participants, identifiable patient data, or animal subjects, and therefore did not require additional ethics-committee approval or informed consent. [Author to confirm consistency with target-journal policy.]
Use of AI-assisted tools.
AI-assisted tools were used to support independent verification of factual claims against peer-reviewed and official sources, and to assist with drafting and formatting. All sources were checked against primary peer-reviewed or official records, and the author takes full responsibility for the content and conclusions. [Disclose per target-journal policy.]
Author contributions.
P.M. conceived the appraisal, performed the analysis and verification, and wrote the manuscript.
References
References are limited to peer-reviewed journal articles and official public-health or publisher records. Non-peer-reviewed materials (preprints, blogs, social media, and news commentary) are excluded.
- Norwitz NG, Feldman D, Soto-Mota A. Seven Years of 700 Cholesterol Without Coronary Atherosclerosis: A Lean Mass Hyper-Responder Case Report. Diseases. 2026;14(5):168. Published 2026 May 11. doi:10.3390/diseases14050168
- Expression of Concern: “Plaque Begets Plaque. ApoB Does Not: Longitudinal Data from The KETO-CTA Trial” [JACC: Advances, Volume 4, Issue 7, July 2025, Article 101686]. JACC Adv. 2026;5(3):102607. doi:10.1016/j.jacadv.2026.102607
- Budoff M, Manubolu VS, Kinninger A, et al. Carbohydrate Restriction-Induced Elevations in LDL-Cholesterol and Atherosclerosis: The KETO Trial. JACC Adv. 2024;3(8):101109. Published 2024 Aug 28. doi:10.1016/j.jacadv.2024.101109
- Nasir K, Mszar R, Cainzos-Achirica M, et al. Age- and sex-based heterogeneity in coronary artery plaque presence and burden in familial hypercholesterolemia: A multi-national study. Am J Prev Cardiol. 2023;17:100611. Published 2023 Nov 23. doi:10.1016/j.ajpc.2023.100611
- Narula J, Stuckey TD, Nakazawa G, et al. Prospective deep learning-based quantitative assessment of coronary plaque by computed tomography angiography compared with intravascular ultrasound: the REVEALPLAQUE study. Eur Heart J Cardiovasc Imaging. 2024;25(9):1287-1295. doi:10.1093/ehjci/jeae115
- Hu P, Dharmayat KI, Stevens CAT, et al. Prevalence of Familial Hypercholesterolemia Among the General Population and Patients With Atherosclerotic Cardiovascular Disease: A Systematic Review and Meta-Analysis. Circulation. 2020;141(22):1742-1759. doi:10.1161/CIRCULATIONAHA.119.044795
- Nordestgaard BG, Chapman MJ, Humphries SE, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013;34(45):3478-90a. doi:10.1093/eurheartj/eht273
- Ference BA, Ginsberg HN, Graham I, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2017;38(32):2459-2472. doi:10.1093/eurheartj/ehx144
- Borén J, Chapman MJ, Krauss RM, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2020;41(24):2313-2330. doi:10.1093/eurheartj/ehz962
- Bolanle IO, de Liedekerke Beaufort GC, Weinberg PD. Transcytosis of LDL Across Arterial Endothelium: Mechanisms and Therapeutic Targets. Arterioscler Thromb Vasc Biol. 2025;45(4):468-480. doi:10.1161/ATVBAHA.124.321549
- ENOS WF, HOLMES RH, BEYER J. Coronary disease among United States soldiers killed in action in Korea; preliminary report. J Am Med Assoc. 1953;152(12):1090-1093. doi:10.1001/jama.1953.03690120006002
- McNamara JJ, Molot MA, Stremple JF, Cutting RT. Coronary artery disease in combat casualties in Vietnam. JAMA. 1971;216(7):1185-1187.
- Strong JP, Malcom GT, McMahan CA, et al. Prevalence and extent of atherosclerosis in adolescents and young adults: implications for prevention from the Pathobiological Determinants of Atherosclerosis in Youth Study. JAMA. 1999;281(8):727-735. doi:10.1001/jama.281.8.727
- Berenson GS, Srinivasan SR, Bao W, Newman WP 3rd, Tracy RE, Wattigney WA. Association between multiple cardiovascular risk factors and atherosclerosis in children and young adults. The Bogalusa Heart Study. N Engl J Med. 1998;338(23):1650-1656. doi:10.1056/NEJM199806043382302
- Cholesterol Treatment Trialists’ (CTT) Collaboration, Baigent C, Blackwell L, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376(9753):1670-1681. doi:10.1016/S0140-6736(10)61350-5
- Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366(9493):1267-1278. doi:10.1016/S0140-6736(05)67394-1
- Cholesterol Treatment Trialists’ Collaboration. Effect of statin therapy on muscle symptoms: an individual participant data meta-analysis of large-scale, randomised, double-blind trials. Lancet. 2022;400(10355):832-845. doi:10.1016/S0140-6736(22)01545-8
- Cheeley MK, Saseen JJ, Agarwala A, et al. NLA scientific statement on statin intolerance: a new definition and key considerations for ASCVD risk reduction in the statin intolerant patient. J Clin Lipidol. 2022;16(4):361-375. doi:10.1016/j.jacl.2022.05.068
- Won KB, Lee SE, Lee BK, et al. Longitudinal assessment of coronary plaque volume change related to glycemic status using serial coronary computed tomography angiography: A PARADIGM (Progression of AtheRosclerotic PlAque DetermIned by Computed TomoGraphic Angiography Imaging) substudy. J Cardiovasc Comput Tomogr. 2019;13(2):142-147. doi:10.1016/j.jcct.2018.12.002
- Sandesara PB, Mehta A, O’Neal WT, et al. Clinical significance of zero coronary artery calcium in individuals with LDL cholesterol ≥190 mg/dL: The Multi-Ethnic Study of Atherosclerosis. Atherosclerosis. 2020;292:224-229. doi:10.1016/j.atherosclerosis.2019.09.014
- Williams MC, Kwiecinski J, Doris M, et al. Low-Attenuation Noncalcified Plaque on Coronary Computed Tomography Angiography Predicts Myocardial Infarction: Results From the Multicenter SCOT-HEART Trial (Scottish Computed Tomography of the HEART). Circulation. 2020;141(18):1452-1462. doi:10.1161/CIRCULATIONAHA.119.044720
- Nieman K, García-García HM, Hideo-Kajita A, et al. Standards for quantitative assessments by coronary computed tomography angiography (CCTA): An expert consensus document of the society of cardiovascular computed tomography (SCCT). J Cardiovasc Comput Tomogr. 2024;18(5):429-443. doi:10.1016/j.jcct.2024.05.232
- Humphries SE, Cooper JA, Seed M, et al. Coronary heart disease mortality in treated familial hypercholesterolaemia: Update of the UK Simon Broome FH register. Atherosclerosis. 2018;274:41-46. doi:10.1016/j.atherosclerosis.2018.04.040
- Centers for Disease Control and Prevention. About Familial Hypercholesterolemia. Atlanta, GA: CDC (official public-health source).
This manuscript is provided for scientific and informational purposes and does not constitute individual medical advice. Clinical decisions should be made with a qualified professional.