
The Interplay of Apolipoprotein B and Systemic Inflammation in Atherosclerotic Cardiovascular Disease: Causality, Mechanisms, and Clinical Paradigms
Atherosclerotic cardiovascular disease (ASCVD) remains the leading cause of morbidity and mortality worldwide. For decades, the field debated the fundamental pathophysiological nature of atherogenesis. Early formulations of the lipid hypothesis viewed atherosclerosis largely as a disease of cholesterol accumulation within the arterial wall, whereas subsequent pathological observations highlighting macrophages, T cells, and other inflammatory cells within plaques fostered the competing view that ASCVD was primarily a chronic inflammatory disorder. Advances in vascular biology, genetics, and clinical trial science now show that these are not mutually exclusive explanations but interlocking components of the same disease process.[1,12,13,22]
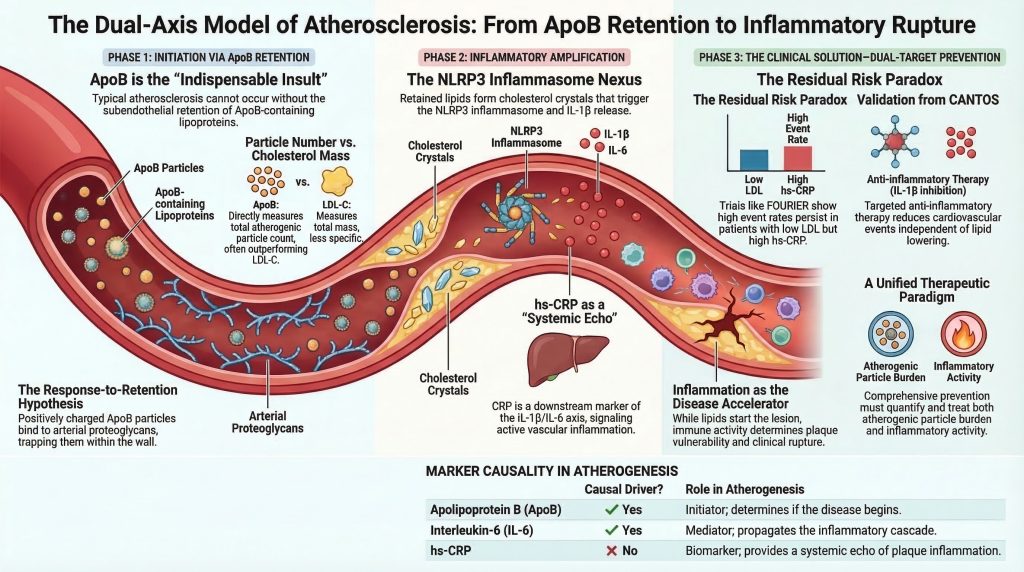
Current scientific and clinical consensus characterizes atherosclerosis as a lipid-driven inflammatory disease. Within this model, apolipoprotein B (ApoB)-containing lipoproteins provide the indispensable initiating insult, while systemic and local inflammation amplifies plaque growth, vulnerability, and rupture risk. Put differently, the lesion begins with retention of atherogenic ApoB particles and becomes dangerous through a maladaptive immune response to retained and modified lipid.[2-5,12,13,21,22]
This review examines the relationship between ApoB, inflammation, and ASCVD progression. By tracing the causal sequence of plaque initiation, reviewing the molecular pathway from lipid retention to cytokine activation, and integrating human genetics with major cardiovascular outcomes trials, it develops a unified model of atherogenesis. The central thesis is that ApoB burden determines whether disease begins, while inflammatory biology strongly influences the pace of progression and the likelihood of myocardial infarction, stroke, or cardiovascular death.[2-5,6-9,12-18,21]
The Causal Sequence: The Response-to-Retention Hypothesis
To understand the relationship between ApoB and inflammation, the temporality of the initial atherogenic event must be established. The key mechanistic question is whether elevated ApoB-containing lipoproteins initiate atherosclerosis before localized inflammation develops, or whether inflammation first primes the arterial wall and subsequently permits lipid deposition. Current evidence strongly favors a sequence in which disease begins with arterial retention of ApoB particles, with inflammation arising as a biological response to those retained particles and then feeding forward to worsen disease.[2-5]
Histopathological and experimental evidence supports the response-to-retention hypothesis of early atherogenesis. In the classic formulation by Williams and Tabas, the influx and trapping of cholesterol-rich ApoB particles within the arterial intima are the necessary first steps in lesion formation. The later Circulation update on subendothelial lipoprotein retention states directly that the initiating process in atherogenesis is retention of ApoB-containing lipoproteins beneath the endothelium, which then triggers the macrophage- and T-cell-dominated inflammatory response that drives lesion development.[2,3]
This model remains compelling because it integrates arterial biology, extracellular matrix binding, and clinical observation. The ApoB particle is not merely a passive lipid carrier. ApoB-containing particles bind arterial wall proteoglycans, especially in vascular regions predisposed to lesion formation. Once trapped, they are exposed to oxidative, enzymatic, and aggregative modification, which converts them into potent inflammatory stimuli. Thus, the earliest event is not generalized inflammation or nonspecific endothelial injury, but focal retention of atherogenic lipoproteins in a susceptible arterial microenvironment.[2-5]
Refuting the “Inflammation-First” Paradigm
Alternative models, most notably the response-to-injury hypothesis, proposed that endothelial injury or pre-existing inflammation must precede lipid deposition. This view gained traction because inflammatory cells are abundant in established plaques and because systemic inflammatory conditions accelerate ASCVD. Yet inflammatory acceleration does not prove inflammatory initiation. The decisive question is whether inflammation, in the absence of sufficient ApoB exposure, can generate a typical cholesterol-rich atheromatous lesion. The balance of evidence suggests that it cannot.[1-3,12,21,22]
Experimental and pathological data indicate that endothelial dysfunction, altered shear stress, and inflammatory activation create a permissive environment for lesion formation, but ApoB burden determines whether lesions actually form and how extensively they progress. Disturbed flow helps explain lesion localization; retained ApoB-containing lipoproteins explain lesion initiation and expansion.[2-5,12,22]
Systemic inflammation nevertheless acts as a powerful disease amplifier. Chronic inflammatory states increase endothelial activation, alter vascular signaling, and accelerate plaque progression. However, in a lipid-poor environment they do not generate the classic cholesterol-laden lesion that defines atherosclerosis. This distinction is clinically important because it explains why anti-inflammatory therapy does not eliminate the need for intensive ApoB lowering.[1,12,13,21,22]
Apolipoprotein B: The Initiating Variable
ApoB exists in 2 major atherogenic isoforms: ApoB48 and ApoB100. ApoB48 is synthesized in the intestine and is present in chylomicrons and their remnants. ApoB100 is synthesized in the liver and is present in very-low-density lipoproteins, intermediate-density lipoproteins, low-density lipoproteins, and lipoprotein(a) [Lp(a)]. Because each atherogenic particle contains exactly 1 ApoB molecule, plasma ApoB concentration reflects the total number of circulating atherogenic particles. This gives ApoB important conceptual and clinical advantages over LDL-C, which reflects cholesterol mass rather than particle number.[4,15,23,25,28]
The clinical importance of ApoB lies in the fact that atherosclerosis is driven by the number of particles capable of entering and being retained in the arterial wall, not simply the amount of cholesterol they carry. Cholesterol content per particle varies considerably, especially in insulin resistance, metabolic syndrome, type 2 diabetes, and hypertriglyceridemia. In such states, LDL-C may underestimate the true burden of atherogenic particles, whereas ApoB captures that burden more directly.[4,23,25,26,28]
Entry of these particles into the arterial wall is driven by the concentration gradient from plasma into the intima. Once inside the subendothelial space, structural domains on ApoB facilitate retention by arterial proteoglycans. Retention prolongs particle residence time and creates the substrate for oxidation, aggregation, and immune activation. Without retention, the downstream inflammatory cascade does not develop in its canonical form.[2-5]
Lp(a) deserves special mention within the ApoB family because it combines an LDL-like ApoB-containing particle with apolipoprotein(a), and it appears to contribute both proatherogenic and proinflammatory effects. Genetic and clinical evidence now support Lp(a) as a causal ASCVD risk factor.[15]
Mechanistic Pathways: From Arterial Retention to Systemic Inflammation
Following subendothelial retention of ApoB-containing particles, a coordinated series of biological events transforms focal lipid accumulation into an inflammatory lesion. This pathway links hyperlipidemia to innate immune activation and explains how a clinically silent fatty streak can evolve into a dangerous plaque.[2,5,10-14,21,22]
Lipoprotein modification
Once retained in the intimal extracellular matrix, ApoB-containing lipoproteins are exposed to oxidative and enzymatic modification. These processes generate oxidized LDL, oxidized phospholipids, and aggregated particles with biological properties distinct from native lipoproteins. Such modified particles act as danger-associated signals within the vessel wall and alter endothelial and myeloid-cell behavior.[2,5,10-14]
These modified lipids are not passive cargo. They become inflammatory ligands that engage vascular and immune cells and sustain lesion evolution. Lp(a), because of its oxidized phospholipid burden, may function as an especially potent inflammatory vehicle after retention.[5,13,15]
Endothelial dysfunction and monocyte recruitment
Modified lipids interact with pattern-recognition pathways and activate inflammatory transcriptional programs in endothelial cells and macrophages. This suppresses atheroprotective endothelial signaling and promotes expression of leukocyte adhesion molecules and chemokines, marking the transition from silent lipid retention to active vascular inflammation.[12-14,21,22]
The result is recruitment of circulating monocytes and T lymphocytes into the subendothelial space. Monocytes adhere, migrate across the activated endothelium, and enter a lipid-rich microenvironment that favors differentiation into macrophages. This is the pivotal handoff from a biochemical lesion driven by lipoprotein retention to a cellular lesion dominated by immune effectors.[12-14,21,22]
Macrophage differentiation, foam cell formation, and crystal genesis
Within the intima, monocytes differentiate into macrophages and internalize modified lipoproteins through scavenger receptors. Because these uptake pathways are not adequately downregulated by intracellular cholesterol loading, macrophages continue to engulf lipid and become foam cells, the hallmark of the early fatty streak.[10-14]
As lipid uptake outpaces cholesterol efflux, intracellular free cholesterol accumulates and can precipitate into cholesterol crystals. These crystals are now recognized not as inert debris, but as inflammatory triggers that activate innate immune pathways central to plaque progression.[10,11,14]
The NLRP3 Inflammasome and Pyroptosis
The intracellular formation and phagocytosis of cholesterol crystals represent a key mechanistic link between retained lipid and innate immune activation. When macrophages attempt to process cholesterol crystals, lysosomal disruption and associated cellular stress activate the NLRP3 inflammasome, a multiprotein danger-sensing complex implicated in atherogenesis.[10,11,14]
Once assembled, NLRP3 activates caspase-1, which cleaves pro-IL-1β and pro-IL-18 into their mature forms and promotes inflammatory cell death pathways. This leads to release of cytokines, proteases, and lipid contents into the extracellular space, contributing to necrotic core expansion and plaque destabilization. This is the point at which chronic lipid-storage lesions become actively destabilizing inflammatory lesions.[10,11,14]
This inflammasome-centered model is therapeutically important because it links retained lipid to IL-1β production and provides a mechanistic rationale for interventions such as canakinumab and colchicine.[6,10,11,14,29-32]
The Hepatic Synthesis of C-Reactive Protein
IL-1β release within the plaque amplifies inflammation both locally and systemically. IL-1β stimulates production of interleukin-6 (IL-6) and other downstream mediators, and IL-6 in turn induces hepatic synthesis of C-reactive protein. Elevated hs-CRP is therefore best understood as a systemic marker of inflammatory signaling arising, at least in part, from inflamed atherosclerotic lesions.[6,12,14,17,18,21,29]
Because CRP sits downstream of the IL-1β/IL-6 axis, it is clinically valuable as a biomarker of inflammatory activity but may not itself be the causal driver of disease.[17,18,21]
The Role of CRP: Biomarker Versus Causal Mediator
Given the strong epidemiologic association between elevated hs-CRP and incident ASCVD, investigators asked whether CRP merely reflects vascular inflammation or directly contributes to atherogenesis. If CRP were causal, CRP itself would be an attractive therapeutic target. If it were only a marker, targeting CRP without affecting upstream pathways would be unlikely to reduce events.[17,18,21]
Evidence from Mendelian randomization
Mendelian randomization has been central to resolving this issue. Genetic studies of lifelong differences in CRP have generally shown null or near-null associations with coronary disease, whereas Mendelian randomization analyses of IL-6 signaling support pathway causality. This pattern strongly suggests that CRP is a downstream biomarker rather than the primary causal mediator.[17,18]
The IL6R Mendelian randomisation analysis is especially informative. Genetic downregulation of IL-6 receptor signaling is associated with lower inflammatory activity, lower downstream CRP, and lower coronary risk, supporting IL-6 pathway causality. This interpretation aligns with later clinical trial data showing benefit from IL-1β inhibition and with the broader view that upstream inflammatory circuitry, not CRP itself, is the relevant therapeutic target.[6,17,18,21]
Evidence from animal models regarding direct CRP effects has been mixed, especially compared with the more consistent genetic and clinical data implicating IL-1β and IL-6. On balance, CRP is best regarded as a high-value clinical biomarker rather than a central therapeutic target.[17,18,21]
Interaction Between ApoB and Inflammation: Landmark Clinical Evidence
Recognition that atherosclerosis depends on both ApoB accumulation and inflammatory amplification led to the modern concept of dual residual risk pathways. Some patients remain at high risk because their atherogenic particle burden remains inadequately controlled. Others achieve major lipid lowering but retain substantial inflammatory risk, reflected by elevated hs-CRP or persistent cytokine activation. Landmark trials such as JUPITER, CANTOS, and FOURIER illustrate these complementary pathways.[6-9,29,33]
JUPITER: inflammation in primary prevention
JUPITER enrolled apparently healthy individuals with LDL-C below 130 mg/dL and hs-CRP of at least 2.0 mg/L, thereby selecting a population with modest traditional lipid levels but increased inflammatory risk. Rosuvastatin reduced both LDL-C and hs-CRP and significantly lowered major cardiovascular events. The trial showed that clinically meaningful cardiovascular risk can exist despite “normal” LDL-C when inflammatory burden is elevated, and that a therapy with both lipid-lowering and anti-inflammatory effects can substantially reduce risk.[7]
CANTOS: isolating the inflammatory hypothesis
CANTOS directly tested whether inflammation reduction independent of lipid lowering improves outcomes. In patients with prior myocardial infarction and persistent hs-CRP elevation despite standard therapy, canakinumab substantially reduced IL-6 and hs-CRP without lowering LDL-C, HDL-C, or ApoB, and reduced recurrent cardiovascular events. Secondary analyses suggested greater benefit in those achieving deeper hs-CRP reduction. CANTOS therefore provided proof of principle that targeted anti-inflammatory therapy can reduce ASCVD events even when lipid levels remain unchanged.[6,29]
FOURIER: persistent inflammatory risk despite ultra-low lipid levels
FOURIER tested intensive lipid lowering with evolocumab in patients with established ASCVD. Evolocumab reduced LDL-C dramatically, while hs-CRP remained essentially unchanged. The trial confirmed benefit from deeper lipid lowering, but subsequent analyses demonstrated that hs-CRP and LDL-C remained independent predictors of outcomes. Even at very low LDL-C levels, higher hs-CRP identified higher residual risk. FOURIER thus showed that residual inflammatory risk persists even when lipid risk is driven to very low levels.[8,9,33]
Together, these trials validate a dual-axis model. JUPITER highlighted inflammatory risk in primary prevention, CANTOS showed that selective anti-inflammatory therapy reduces events without lipid lowering, and FOURIER showed that intense lipid lowering leaves a measurable pool of residual inflammatory risk. The implication is not to choose between lipid and inflammation, but to treat both.[6-9,29,33]
Epidemiological Synergy: Insights From UK Biobank and Contemporary Cohorts
Large population analyses complement trial data by showing how ApoB and inflammatory risk interact across broad risk distributions. Contemporary cohort studies and UK Biobank-based analyses indicate that ApoB burden and inflammatory markers often provide complementary information, with the highest event rates often observed when both are elevated. Particularly important are discordance analyses showing that ApoB often predicts risk better than LDL-C when the 2 measures disagree.[4,23,25,28]
In metabolic syndrome and hypertriglyceridemia, LDL-C may appear deceptively low because each particle carries less cholesterol, whereas ApoB still reflects the true number of atherogenic particles. Recent cohort work also suggests that ApoB-based approaches can improve risk discrimination relative to cholesterol-based metrics in discordant states.[4,23,25,26,28]
These epidemiologic observations reinforce the mechanistic framework. Elevated ApoB indicates increased opportunity for arterial entry and retention, whereas elevated hs-CRP signals active inflammatory amplification. When both are present, risk rises materially. That does not place CRP on the same causal level as ApoB, but it does support measuring both to refine clinical risk assessment.[4,17,18,23,25,28]
Clinical Implications and 2025 Guideline Perspectives
Modern lipidology increasingly recognizes that because cholesterol mass per LDL particle varies, reliance on total cholesterol or calculated LDL-C alone can misclassify risk. ApoB directly inventories the circulating atherogenic particles capable of entering the arterial wall. The 2024 Circulation review on ApoB argues for broader clinical use of ApoB because of its stronger biologic alignment with ASCVD causality and its utility in discordant states.[4,23,25,28]
This shift is reflected in contemporary guidance. The 2025 AACE dyslipidemia guideline emphasizes modern risk-based pharmacologic management, while the 2025 focused ESC/EAS update supports earlier and broader use of combination therapy, including statins, ezetimibe, bempedoic acid, and PCSK9 inhibition, to reduce atherogenic burden. Although society-specific thresholds differ, the overall direction is consistent: ApoB is gaining prominence as an actionable marker, especially in high-risk and discordant patients.[19,20]
Inflammation has likewise moved from an academic concept to a clinically actionable one. The 2025 ACC scientific statement emphasizes the clinical relevance of inflammation across the ASCVD continuum. hs-CRP remains useful for risk enhancement in selected primary prevention settings and for identifying residual inflammatory risk in secondary prevention. Low-dose colchicine has emerged as a practical anti-inflammatory option in selected patients with established ASCVD, and ongoing work on IL-6 pathway modulation and Lp(a)-lowering strategies may further refine treatment.[20,27,30-32,35]
Unresolved Scientific Debates: Defining the Disease
Despite broad convergence, semantic debate persists over whether atherosclerosis is fundamentally lipid driven, inflammatory, or lipid-induced inflammatory. The lipid-centric view is correct in one crucial sense: without ApoB-containing lipoproteins, typical atherosclerosis does not arise. Human genetics, pathology, and lipid-lowering interventions all strongly support this point.[2-5,15,16,23]
Yet a purely lipid-centric model does not fully explain why some patients with excellent lipid control continue to experience plaque rupture and recurrent events. Conversely, the inflammation-centric view correctly emphasizes that immune mechanisms dominate plaque destabilization, cap degradation, and rupture, but inflammation in a lipid-poor environment does not produce the classic cholesterol-rich atheroma. The most accurate synthesis is therefore that atherosclerosis is a lipid-induced inflammatory disease: initiated by ApoB retention, then amplified and rendered clinically dangerous by maladaptive innate and adaptive immune responses to retained lipid.[1-3,10-14,21,22,27,29-35]
Conclusion
The pathophysiology of ASCVD requires convergence of 2 closely linked biological axes. Disease begins with influx and subendothelial retention of ApoB-containing lipoproteins within the arterial wall. This establishes the initiating lesion. Progression from a clinically silent fatty streak to a vulnerable, necrotic, rupture-prone plaque depends on inflammatory amplification driven by modified lipids, foam-cell biology, cholesterol crystals, NLRP3 inflammasome activation, and cytokine signaling through IL-1β and IL-6. hs-CRP emerges from this cascade as an informative systemic marker rather than the primary causal agent.[2-5,10-14,17,18,21]
Landmark trials over the past 2 decades have shown that treating the ApoB driver through statins and PCSK9 inhibition and treating the inflammatory amplifier through IL-1β inhibition or colchicine can provide independent and complementary benefit. To reduce residual cardiovascular risk meaningfully, contemporary practice must move beyond cholesterol mass alone and assess both atherogenic particle burden and residual inflammatory activity. That dual-risk framework is the clearest practical implication of modern atherosclerosis research.[4,6-9,19,20,27,29-35]
References
- Libby P. Inflammation in atherosclerosis. Nature. 2002;420(6917):868-874. doi:10.1038/nature01323
- Tabas I, Williams KJ, Borén J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications.
- Williams KJ, Tabas I. The response-to-retention hypothesis of early atherogenesis.
- De Oliveira-Gomes D, Joshi PH, Peterson ED, Rohatgi A, Khera A, Navar AM. Apolipoprotein B: Bridging the Gap Between Evidence and Clinical Practice. Circulation. 2024;150(1):62-79. doi:10.1161/CIRCULATIONAHA.124.068885
- Borén J, Williams KJ. The central role of arterial retention of cholesterol-rich apolipoprotein-B-containing lipoproteins in the pathogenesis of atherosclerosis: a triumph of simplicity. Curr Opin Lipidol. 2016;27(5):473-483. doi:10.1097/MOL.0000000000000330
- Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med. 2017;377(12):1119-1131. doi:10.1056/NEJMoa1707914
- Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359(21):2195-2207. doi:10.1056/NEJMoa0807646
- Sabatine MS, Giugliano RP, Keech AC, et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N Engl J Med. 2017;376(18):1713-1722. doi:10.1056/NEJMoa1615664
- Pradhan AD, Aday AW, Rose LM, Ridker PM. Residual Inflammatory Risk on Treatment With PCSK9 Inhibition and Statin Therapy. Circulation. 2018;138(2):141-149. doi:10.1161/CIRCULATIONAHA.118.034645
- Duewell P, Kono H, Rayner KJ, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464(7293):1357-1361. doi:10.1038/nature08938
- Grebe A, Latz E. Cholesterol crystals and inflammation. Curr Rheumatol Rep. 2013;15(3):313. doi:10.1007/s11926-012-0313-z
- Libby P, Ridker PM, Hansson GK; Leducq Transatlantic Network on Atherothrombosis. Inflammation in atherosclerosis: from pathophysiology to practice. J Am Coll Cardiol. 2009;54(23):2129-2138. doi:10.1016/j.jacc.2009.09.009
- Tall AR, Yvan-Charvet L. Cholesterol, inflammation and innate immunity. Nat Rev Immunol. 2015;15(2):104-116. doi:10.1038/nri3793
- Grebe A, Hoss F, Latz E. NLRP3 Inflammasome and the IL-1 Pathway in Atherosclerosis. Circ Res. 2018;122(12):1722-1740. doi:10.1161/CIRCRESAHA.118.311362
- Tsimikas S, Stroes ESG. The dedicated “Lp(a) clinic”: A concept whose time has arrived?. Atherosclerosis. 2020;300:1-9. doi:10.1016/j.atherosclerosis.2020.03.003
- Ference BA, Kastelein JJP, Ray KK, et al. Association of Triglyceride-Lowering LPL Variants and LDL-C-Lowering LDLR Variants With Risk of Coronary Heart Disease. JAMA. 2019;321(4):364-373. doi:10.1001/jama.2018.20045
- Zacho J, Tybjaerg-Hansen A, Jensen JS, Grande P, Sillesen H, Nordestgaard BG. Genetically elevated C-reactive protein and ischemic vascular disease. N Engl J Med. 2008;359(18):1897-1908. doi:10.1056/NEJMoa0707402
- Interleukin-6 Receptor Mendelian Randomisation Analysis (IL6R MR) Consortium, Swerdlow DI, Holmes MV, et al. The interleukin-6 receptor as a target for prevention of coronary heart disease: a mendelian randomisation analysis. Lancet. 2012;379(9822):1214-1224. doi:10.1016/S0140-6736(12)60110-X
- Mach F, Koskinas KC, Roeters van Lennep JE, et al. 2025 Focused Update of the 2019 ESC/EAS Guidelines for the management of dyslipidaemias. Eur Heart J. 2025;46(42):4359-4378. doi:10.1093/eurheartj/ehaf190
- Patel SB, Wyne KL, Afreen S, et al. American Association of Clinical Endocrinology Clinical Practice Guideline on Pharmacologic Management of Adults With Dyslipidemia. Endocr Pract. 2025;31(2):236-262. doi:10.1016/j.eprac.2024.09.016
- Ridker PM. From C-Reactive Protein to Interleukin-6 to Interleukin-1: Moving Upstream To Identify Novel Targets for Atheroprotection. Circ Res. 2016;118(1):145-156. doi:10.1161/CIRCRESAHA.115.306656
- Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol. 2011;12(3):204-212. doi:10.1038/ni.2001
- Sniderman AD, Thanassoulis G, Glavinovic T, et al. Apolipoprotein B Particles and Cardiovascular Disease: A Narrative Review. JAMA Cardiol. 2019;4(12):1287-1295. doi:10.1001/jamacardio.2019.3780
- Aday AW, Ridker PM. Targeting Residual Inflammatory Risk: A Shifting Paradigm for Atherosclerotic Disease. Front Cardiovasc Med. 2019;6:16. Published 2019 Feb 28. doi:10.3389/fcvm.2019.00016
- Mora S, Buring JE, Ridker PM. Discordance of low-density lipoprotein (LDL) cholesterol with alternative LDL-related measures and future coronary events. Circulation. 2014;129(5):553-561. doi:10.1161/CIRCULATIONAHA.113.005873
- Arsenault BJ, Després JP, Stroes ES, et al. Lipid assessment, metabolic syndrome and coronary heart disease risk. Eur J Clin Invest. 2010;40(12):1081-1093. doi:10.1111/j.1365-2362.2010.02357.x
- Mensah GA, Arnold N, Prabhu SD, Ridker PM, Welty FK. Inflammation and Cardiovascular Disease: 2025 ACC Scientific Statement: A Report of the American College of Cardiology. J Am Coll Cardiol. Published online September 29, 2025. doi:10.1016/j.jacc.2025.08.047
- Sniderman AD, Dufresne L, Pencina KM, Bilgic S, Thanassoulis G, Pencina MJ. Discordance among apoB, non-high-density lipoprotein cholesterol, and triglycerides: implications for cardiovascular prevention. Eur Heart J. 2024;45(27):2410-2418. doi:10.1093/eurheartj/ehae258
- Ridker PM, MacFadyen JG, Everett BM, et al. Relationship of C-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: a secondary analysis from the CANTOS randomised controlled trial. Lancet. 2018;391(10118):319-328. doi:10.1016/S0140-6736(17)32814-3
- Nidorf SM, Fiolet ATL, Mosterd A, et al. Colchicine in Patients with Chronic Coronary Disease. N Engl J Med. 2020;383(19):1838-1847. doi:10.1056/NEJMoa2021372
- Nidorf SM, Eikelboom JW, Budgeon CA, Thompson PL. Low-dose colchicine for secondary prevention of cardiovascular disease. J Am Coll Cardiol. 2013;61(4):404-410. doi:10.1016/j.jacc.2012.10.027
- Tardif JC, Kouz S, Waters DD, et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N Engl J Med. 2019;381(26):2497-2505. doi:10.1056/NEJMoa1912388
- O’Donoghue ML, Fazio S, Giugliano RP, et al. Lipoprotein(a), PCSK9 Inhibition, and Cardiovascular Risk. Circulation. 2019;139(12):1483-1492. doi:10.1161/CIRCULATIONAHA.118.037184
- Ference BA, Kastelein JJP, Ginsberg HN, et al. Association of Genetic Variants Related to CETP Inhibitors and Statins With Lipoprotein Levels and Cardiovascular Risk. JAMA. 2017;318(10):947-956. doi:10.1001/jama.2017.11467
- Du Z, Li F, Jiang L, et al. Metabolic systems approaches update molecular insights of clinical phenotypes and cardiovascular risk in patients with homozygous familial hypercholesterolemia. BMC Med. 2023;21(1):275. Published 2023 Jul 27. doi:10.1186/s12916-023-02967-8