
Understanding Your Heart: High Cholesterol and Your Family History
1. The Big Picture: How Your Heart Stays Healthy
Think of your heart as the most important pump in your home, and your arteries as the “pipes” that carry life-sustaining blood to every room in your body. For the pump to work perfectly, those pipes need to stay clear and open. To check on the health of your plumbing, doctors look at “lipids”—special fats and proteins in your blood—to see if the pipes are at risk of getting clogged.The central goal of this summary is to solve a medical mystery: why does having high cholesterol matter even if everyone in your family has a healthy heart? Many people believe that if their parents and grandparents never had heart trouble, they are naturally “immune” to high cholesterol. However, by looking at how “sticky stuff” builds up in the blood, we can see that a good family history might give you a head start, but it isn’t a permanent shield against future clogs.
2. Meet the “Sticky Particles”: LDL-C and ApoB
Not everything floating in your blood is the same. Some particles are harmless, while others act like “trash” that can get stuck inside your artery walls. When doctors measure your risk, they look at two main markers:
- High LDL-C (The Weight): This measures the total “weight” of bad cholesterol. A level of 190 mg/dL or higher is a major warning sign.
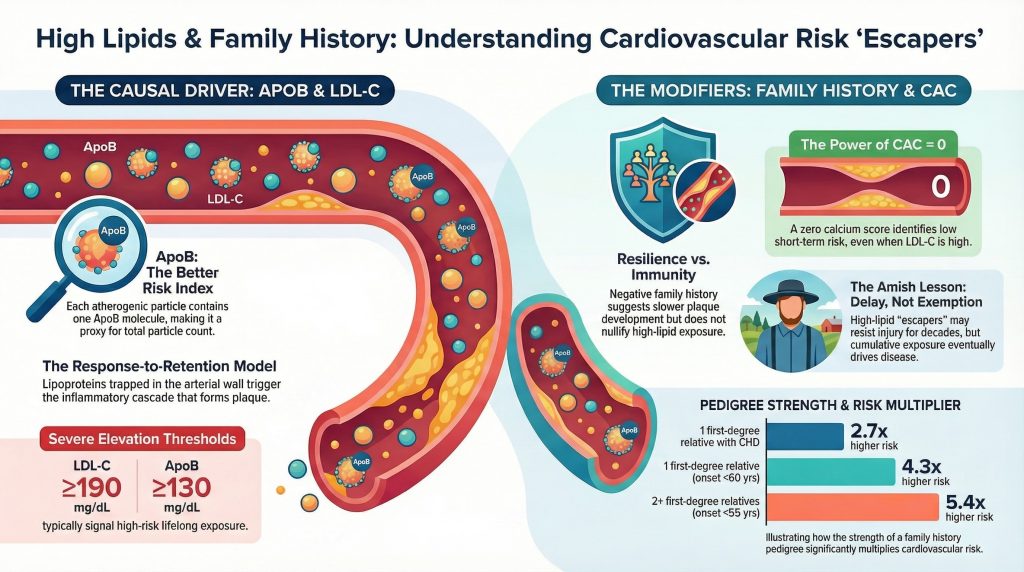
- High ApoB (The Count): This is often a “better index” of risk. Imagine a highway: LDL-C is the total weight of all the trucks, but ApoB counts the actual number of cars on the road. Because every sticky particle has exactly one ApoB molecule, counting ApoB tells doctors exactly how much “trash” is threatening to clog the pipes. A level of 130 mg/dL or higher is considered high.Science Fact: The “Response-to-Retention” Model Why are these particles considered “causal”? It’s because of how they behave. They don’t just flow past the artery walls; they actually get trapped inside the walls. Once they are stuck, they trigger an alarm in the body, leading to the growth of “plaque”—the gunk that eventually causes heart attacks.
3. The “Family Shield”: Is It a Superpower?
Scientists often hunt for “escapers”—people who have very high cholesterol but no history of heart disease in their family. Doctors call this “Vascular Resilience.” It means the person’s pipes are temporarily better at ignoring the gunk. Their artery walls might be less “leaky,” making it harder for the sticky particles to get trapped inside.However, a healthy family history can be falsely reassuring . You must consider two things:
- The Statin Secret: Your parents might have “healthy” hearts only because they are taking modern medicines (like statins) to keep their pipes clear.
- The Size Factor: If your family tree is small, there might not be enough people to show a pattern of heart trouble yet.Having a resilient body can delay the start of heart problems, but it does not abolish the danger. Resilience is like having a sturdy umbrella in a rainstorm—it helps for a while, but if the storm never ends, you’re eventually going to get wet.
4. Lessons from the Amish: A Real-Life Example
To see if a healthy lifestyle and strong family genes can truly “cancel out” high cholesterol, scientists studied a specific community of Amish people in Pennsylvania. Many people in this group have a “genetic glitch” (known as the ApoB R3500Q founder variant ) that gives them very high cholesterol from birth.In the Amish study, the children and young adults looked perfectly healthy. Their resilient bodies were resisting the high cholesterol for decades. But the “So What?” comes later: by the time these individuals reached middle age , they were 4.5 times more likely to have clogs in their hearts than people with normal cholesterol. This proves that high cholesterol is a relentless pressure . Even if you have “super-pipes,” the constant trapping of particles eventually catches up to the heart.
5. The “Heart Camera”: Using the CAC Test
Rather than guessing based on your family tree, doctors can use a “heart camera” called a Coronary Artery Calcium (CAC) test. This scan looks directly at the pipes to see if any hard clogs have formed.Doctors look for the “Power of Zero.” A score of 0 means no hard clogs are visible right now . While this is great news for your short-term safety, it has limits:
- The “Right Now” vs. “Lifetime” Gap: A zero means you are safe today, but it doesn’t mean you’ll be safe in ten years if your ApoB stays high.
- Hidden Soft Gunk: The camera sees “old,” hard clogs but can miss “new,” soft gunk that hasn’t hardened yet.Because the damage from cholesterol adds up over time, doctors usually suggest starting medicine for anyone with an LDL-C over 190, even if their “heart camera” shows a zero today. They want to stop the “trapping” process before the first clog ever forms.
6. Final Summary: Resilience vs. Risk
In the end, a healthy family history is a modifier (a helper) but not a shield (total protection). High ApoB and LDL-C are the main drivers of heart trouble. Ignoring them because your family has healthy hearts is like ignoring a leak in your own plumbing just because the neighbors’ pipes are fine.Your family history might mean your body is more resilient, but the underlying biology of high lipids is a lifelong challenge. By working with doctors to monitor your “particle count” and using tools like the CAC test, you can take charge of your health. Remember: it’s much easier to keep a pipe clean than it is to fix a clog once it has already formed.
DEEP DIVE
Family History vs High Lipids
The Paradox of Lipid-Driven Risk: Evaluating the Protective Capacity of Negative Family History in Individuals With High LDL-C and ApoB
The contemporary model of atherosclerotic cardiovascular disease (ASCVD) holds that circulating apolipoprotein B (ApoB)-containing lipoproteins are causal in atherogenesis and that risk is strongly related to both the magnitude and duration of exposure. In this framework, low-density lipoprotein cholesterol (LDL-C) is clinically important, but ApoB is often more mechanistically informative because each atherogenic particle contains a single ApoB molecule, making ApoB a practical proxy for total atherogenic particle number. Adults with LDL-C levels of 190 mg/dL or greater are classified as having severe hypercholesterolemia and are typically evaluated for possible familial hypercholesterolemia (FH), especially when accompanied by tendon xanthomas, a suggestive pedigree, or persistently marked elevation from early life. ApoB values of 130 mg/dL or greater are generally treated as risk-enhancing, but they are not by themselves diagnostic of monogenic FH.1,2 (Lipid Journal)
Yet clinical practice repeatedly reveals a puzzling subgroup of “escapers”: individuals with very high LDL-C or ApoB who remain free of myocardial infarction, clinically manifest coronary disease, or detectable coronary calcification well into midlife or later. This creates a central question in preventive cardiology: can a strongly negative family history of coronary disease, especially one spanning multiple generations without premature events, meaningfully offset the expected hazard associated with high ApoB exposure? The most defensible answer is nuanced. Negative family history appears to act as a favorable risk modifier and may identify individuals with slower plaque development or greater vascular resilience, but it does not nullify the biologic effect of lifelong exposure to atherogenic particles. High ApoB remains causally upstream; family history modifies the expression of that burden rather than abolishing it.1-3 (Lipid Journal)
Accordingly, the relevant clinical issue is not whether a negative pedigree proves that high LDL-C is harmless. Rather, it is whether favorable inherited biology, as reflected imperfectly by family history, can delay the onset of plaque, reduce the inflammatory consequences of retained lipoproteins, and produce a sufficiently low short-term event rate to justify more individualized management. That possibility is real, but it must be evaluated alongside direct evidence of disease, especially coronary artery calcium (CAC), rather than family history alone.1,2,4 (Lipid Journal)
The Mechanistic Primacy of ApoB in Atherogenesis
The pathobiology of atherosclerosis is best understood through the response-to-retention model. ApoB-containing lipoproteins cross the endothelial barrier, enter the arterial intima, and become retained through interactions with arterial-wall proteoglycans. This retained lipoprotein material undergoes modification, which promotes endothelial activation, monocyte recruitment, macrophage infiltration, foam-cell formation, smooth-muscle migration, necrotic core development, and eventually plaque progression and calcification. In this sequence, plasma cholesterol concentration matters chiefly because it reflects the amount of cholesterol carried by particles capable of entering and being trapped in the vessel wall. ApoB is often the better index of this burden because it measures the number of circulating atherogenic particles rather than the cholesterol mass within them.1 (Lipid Journal)
This distinction is particularly important when LDL-C and ApoB are discordant. A person can have relatively cholesterol-poor but numerous LDL particles, resulting in a higher particle burden than LDL-C alone suggests. Conversely, another individual may have fewer, cholesterol-richer particles. For this reason, ApoB tends to improve risk discrimination in settings such as metabolic syndrome, hypertriglyceridemia, and insulin resistance, where LDL-C may understate true atherogenic exposure. Nevertheless, LDL-C remains clinically indispensable, especially because current treatment guidelines and most outcomes trials still use LDL-C thresholds and targets.1,2 (Lipid Journal)
If lifelong elevation of LDL-C and ApoB is the core driver of atherogenesis, then the biologic basis for apparent protection must lie not in dismissal of the lipid hypothesis but in modulation of downstream steps. A patient with a negative family history despite high LDL-C may have inherited a vessel wall that is less permeable to particle entry, less adhesive to monocytes, less prone to inflammatory amplification, or less vulnerable to thrombosis and plaque rupture. In other words, family history may encode differences in susceptibility rather than differences in exposure.1,5 (Lipid Journal)
Genetic Models of Protection and Attenuated Risk Expression
Human genetics provides strong proof that inherited biology can markedly modify cardiovascular risk. The clearest example remains PCSK9. PCSK9 promotes degradation of hepatic LDL receptors; loss-of-function variants preserve receptor recycling, lower lifelong LDL-C, and substantially reduce coronary risk. In the classic study by Cohen et al, Black carriers of nonsense variants in PCSK9 had approximately 28% lower LDL-C and an 88% lower risk of coronary heart disease, while White carriers of the R46L variant had approximately 15% lower LDL-C and a 47% lower risk.3 This study established a broader principle that is highly relevant here: small lifelong changes in biologic pathways can produce large lifetime differences in coronary outcomes.3 (New England Journal of Medicine)
PCSK9 demonstrates protection by reducing the causal exposure itself. Other variants suggest that coronary protection can also arise through modulation of inflammation or lipoprotein composition. A Mendelian randomization analysis of the interleukin-6 receptor (IL6R) pathway showed that the Asp358Ala variant was associated with altered inflammatory signaling and a modest reduction in coronary heart disease risk, supporting the view that inflammatory responsiveness contributes causally to plaque progression.5 Likewise, variants affecting APOC3, ANGPTL3, and related pathways can reduce triglyceride-rich remnant burden, potentially lowering residual atherogenic and inflammatory load even when LDL-C remains elevated.1,5 (The Lancet)
These observations matter because they show how two people with the same LDL-C can experience markedly different clinical trajectories. One may carry additional inherited liabilities involving Lp(a), remnant metabolism, inflammation, endothelial function, or thrombosis; another may carry a comparatively resilient vascular phenotype. A strongly negative family history is therefore plausible as a marker of lower inherited vulnerability, but it remains an imprecise marker. It cannot identify the specific protective pathways involved, and it cannot establish whether protection is durable or merely delays disease expression.1,5 (Lipid Journal)
Lessons From the Amish: Delay Rather Than Immunity
The Pennsylvania Amish founder mutation in APOB offers a useful real-world model. Familial defective ApoB due to the ApoB R3500Q founder variant produces lifelong LDL elevation in a population with relatively homogeneous background and distinctive environmental exposures. Studies in Amish children and young adults have shown markedly elevated LDL-C with limited early evidence of vascular injury by carotid intima-media thickness or pulse-wave velocity. This initially appears to support substantial biologic protection or at least a prolonged period of vascular tolerance.6 (Open Access Journals)
However, longer-term follow-up undermines the idea of permanent immunity. In adult carriers, coronary calcification becomes substantially more common. Review data from the Amish literature indicate that middle-aged carriers were approximately 4.5 times more likely than noncarriers to have detectable CAC.6 This is a critical finding. It suggests that protective background biology, favorable environment, or both may delay disease expression, but the cumulative burden of high ApoB exposure still exerts pressure over time. The vessel wall may resist injury for decades, yet the exposure is relentless.6 (Open Access Journals)
The Amish example is therefore directly relevant to the problem of negative family history. A favorable pedigree may reflect inherited resilience, but resilience is not equivalent to exemption. In patients with severe lifelong hypercholesterolemia, the most evidence-based assumption is that apparent protection often represents slower plaque development rather than true freedom from eventual disease.2,6 (AHA Journals)
Quantifying the Effect of Family History
Family history is best understood as a compressed clinical signal that captures shared genetics, shared behaviors, and shared environment. A positive family history of coronary disease, especially if premature, is a well-established risk enhancer. The absence of such a history is favorable, but the degree of protection depends heavily on the pedigree definition. In the Newcastle Family History Study, the estimated odds ratio for an acute coronary event rose as the pedigree became stronger and more premature: approximately 2.7 for at least 1 first-degree relative with coronary heart disease at any age, 4.3 for at least 1 first-degree relative with coronary heart disease before age 60 years, and 5.4 for 2 or more first-degree relatives with coronary heart disease before age 55 years.7 (OUP Academic)
These data support 2 important conclusions. First, family history is not trivial; it conveys substantial information beyond routine lipid measures. Second, the inverse does not justify overcorrection. Someone with LDL-C of 190 mg/dL and no family history is not suddenly risk-equivalent to a person with LDL-C of 90 mg/dL. Rather, that person may be at lower relative risk than another patient with the same LDL-C but an adverse pedigree. Family history modifies the slope of risk expression, not the underlying causal exposure.2,7 (AHA Journals)
More contemporary work similarly shows that family history adds predictive value beyond conventional risk factors. It appears to improve risk assessment not merely because of event clustering, but because it also correlates with earlier development of subclinical disease. Thus, when negative family history is present in a patient with markedly elevated LDL-C or ApoB, it is reasonable to interpret it as reassuring, but only in a bounded sense: it suggests lower inherited susceptibility than expected, not biologic invulnerability.7 (OUP Academic)
Family History, Polygenic Risk, and the Concept of a “Resilient” Vessel Wall
Negative family history likely reflects more than the absence of a single monogenic mutation. It may also indicate a lower aggregate burden of common proatherogenic variants and a relative absence of inherited inflammatory or thrombotic predisposition. Modern thinking about polygenic risk supports the idea that common small-effect variants can meaningfully shape the age at onset and likelihood of disease. Family history and polygenic risk are therefore complementary rather than interchangeable: family history captures observed disease clustering, while polygenic scores estimate inherited burden from measured variants. A patient with high LDL-C but low overall inherited susceptibility may remain event-free substantially longer than one with the same LDL-C plus high polygenic burden.1,7 (Lipid Journal)
Clinically, this helps explain why some patients with severe hypercholesterolemia develop coronary disease very early while others do not. The artery is not responding to ApoB in a vacuum. The burden of particle retention is filtered through endothelial function, inflammatory set-point, thrombogenicity, remnant metabolism, Lp(a), glycemic state, blood pressure, and likely additional pathways not fully captured in routine care. A strongly negative family history is one clue that this broader biologic background may be favorable. But because it is only a clue, it should be interpreted alongside direct markers of disease.1,2 (Lipid Journal)
CAC as a Direct Measure: The Power of Zero
Among available clinical tools, CAC scoring is generally more informative than pedigree alone when the question is whether a patient with high LDL-C is currently expressing coronary atherosclerosis. This is particularly relevant in severe hypercholesterolemia, where traditional lipid-based risk estimation may exaggerate short-term risk for some patients while remaining correct about lifetime hazard. The MESA analysis by Sandesara et al examined individuals with LDL-C of 190 mg/dL or greater and found that a CAC score of 0 identified a subgroup with low short-term event rates.4 (Atherosclerosis Journal)
Secondary reporting of those data cites an event rate of approximately 4.7 per 1000 person-years among those with LDL-C of 190 mg/dL or greater and CAC of 0, with risk roughly 5-fold higher when CAC was present.4 The practical implication is not that severe LDL elevation becomes benign when CAC is absent. Rather, it means that the patient’s current plaque burden is lower than expected, and therefore near-term risk is lower than a lipid value alone would imply. This is exactly the setting in which negative family history becomes most clinically meaningful: it can help explain why a patient with severe hypercholesterolemia nevertheless has no calcified plaque in midlife.4 (Europe PMC)
Still, CAC has limitations. It identifies calcified plaque, not all plaque. Patients with CAC of 0 may still harbor noncalcified plaque, and events can still occur, especially in the presence of other high-risk features. This is why the 2018 AHA/ACC cholesterol guideline states that if CAC is 0, statin therapy may be withheld or delayed in selected primary prevention patients, except in cigarette smokers, those with diabetes mellitus, and those with a strong family history of premature ASCVD.2 That exception matters. Family history retains significance precisely because the absence of calcium does not fully exclude vulnerable biology.2,4 (AHA Journals)
The same guideline also makes clear that LDL-C of 190 mg/dL or greater constitutes severe hypercholesterolemia and generally warrants statin therapy independent of calculated 10-year risk.2 Therefore, CAC and family history should not be used to argue that such patients are genuinely low risk in an absolute sense. They should instead be used to refine short-term risk assessment, pace the intensity of escalation, and support more individualized shared decision-making.2,4 (AHA Journals)
Lp(a), hs-CRP, and Metabolic Context
The argument for relative protection is strongest when negative family history aligns with other favorable biomarkers. Lp(a) is particularly important because it confers both atherogenic and prothrombotic risk independent of standard LDL measures. The National Lipid Association’s updated guidance recommends measuring Lp(a) at least once in every adult for risk stratification; values below 75 nmol/L, or below 30 mg/dL, are considered low risk, whereas values of 125 nmol/L or greater, or 50 mg/dL or greater, are treated as risk-enhancing.8 Thus, a patient with high LDL-C but low Lp(a) lacks one major inherited accelerator of plaque progression and thrombosis.8 (Lipid Journal)
Similarly, high-sensitivity C-reactive protein (hs-CRP) provides a rough index of inflammatory activation. The 2019 ACC/AHA primary prevention guideline lists hs-CRP of 2.0 mg/L or greater as a risk-enhancing factor. A persistently low hs-CRP does not guarantee absence of arterial inflammation, but it is directionally consistent with a less activated vascular milieu. When negative family history coexists with low Lp(a), low hs-CRP, normal blood pressure, normal glycemia, low triglycerides, and CAC of 0, the case for a slower atherosclerotic trajectory becomes much stronger.2,8 (AHA Journals)
Metabolic context also matters. In patients with insulin resistance or hypertriglyceridemia, ApoB can reveal hidden particle excess not captured by LDL-C. Conversely, patients with isolated LDL elevation, low triglycerides, and otherwise favorable metabolic parameters may have a more benign short-term risk phenotype than those with the same LDL-C embedded within broader cardiometabolic dysfunction. Again, the key distinction is between tempo and causality: favorable metabolic context can slow disease expression without making lifelong ApoB elevation innocuous.1,2 (Lipid Journal)
Clinical Interpretation and Limits of the “Protection” Argument
The strongest clinically supportable conclusion is that a strongly negative family history can meaningfully attenuate relative risk among patients with high LDL-C or ApoB, especially when supported by CAC of 0 and other favorable markers. It can justify a less alarmist interpretation of risk and may support more individualized decisions regarding how rapidly to intensify therapy. However, it should not be framed as proof that severe hypercholesterolemia is safe untreated over the life course.2,4,7 (AHA Journals)
There are also important failure points in pedigree interpretation. A “negative family history” may be falsely reassuring if earlier generations were effectively treated with statins or antihypertensives before events occurred. Family size may be too small to be informative. Competing causes of death may censor coronary expression. Shared behavior may masquerade as genetic protection. For these reasons, family history is valuable but never definitive. It should inform judgment, not dominate it.2,7 (AHA Journals)
A more defensible clinical formulation is therefore as follows: in an asymptomatic middle-aged adult with LDL-C of 190 mg/dL or greater, ApoB elevation, strongly negative family history, low Lp(a), excellent metabolic health, and CAC of 0, short-term event risk may be lower than expected for severe hypercholesterolemia. But lifetime risk remains elevated relative to individuals without prolonged ApoB exposure, and serial monitoring remains prudent if treatment intensity is moderated.2,4,8 (AHA Journals)
Conclusion
A strong negative family history of coronary disease appears capable of meaningfully attenuating the clinical expression of lipid-driven risk, but the evidence supports interpreting it as a risk modifier rather than a biologic shield that neutralizes lifelong ApoB exposure. The most plausible explanation is that favorable inherited vascular biology can slow plaque initiation, blunt inflammatory amplification, reduce thrombotic vulnerability, or lower exposure to important co-drivers such as Lp(a) and remnants.1,5,8 (Lipid Journal)
In practical terms, direct measurement of disease, especially CAC, is more informative than pedigree alone for assessing whether this favorable biology has translated into low current plaque burden. Patients with severe hypercholesterolemia and CAC of 0 may indeed have low short-term event rates, particularly when family history is negative and other biomarkers are favorable. But the cumulative burden of high ApoB remains operative in the background, which is why guideline-based care still treats LDL-C of 190 mg/dL or greater as a major lifetime risk state.2,4 The most accurate synthesis is therefore this: negative family history can delay and attenuate the manifestation of high-lipid risk, sometimes substantially, but it does not abolish the underlying biology that makes long-term ApoB exposure dangerous.2,4,7 (AHA Journals)
References
- Soffer DE, Marston NA, Maki KC, et al. Role of apolipoprotein B in the clinical management of cardiovascular risk in adults: An Expert Clinical Consensus from the National Lipid Association. J Clin Lipidol. 2024;18(5):e647-e663. doi:10.1016/j.jacl.2024.08.013
- Grundy SM, Stone NJ, Bailey AL, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;139(25):e1082-e1143. doi:10.1161/CIR.0000000000000625
- Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354(12):1264-1272. doi:10.1056/NEJMoa054013
- Sandesara PB, Mehta A, O’Neal WT, et al. Clinical significance of zero coronary artery calcium in individuals with LDL cholesterol ≥190 mg/dL: The Multi-Ethnic Study of Atherosclerosis. Atherosclerosis. 2020;292:224-229. doi:10.1016/j.atherosclerosis.2019.09.014
- Interleukin-6 Receptor Mendelian Randomisation Analysis (IL6R MR) Consortium, Swerdlow DI, Holmes MV, et al. The interleukin-6 receptor as a target for prevention of coronary heart disease: a mendelian randomisation analysis. Lancet. 2012;379(9822):1214-1224. doi:10.1016/S0140-6736(12)60110-X
- Snyder C, Beitelshees AL, Chowdhury D. Familial Hyperlipidemia Caused by Apolipoprotein B Mutation in the Pediatric Amish Population: A Mini Review. Interv Cardiol (Lond). 2023;15(Suppl 17):433-437.
- Silberberg JS, Wlodarczyk J, Fryer J, Robertson R, Hensley MJ. Risk associated with various definitions of family history of coronary heart disease. The Newcastle Family History Study II. Am J Epidemiol. 1998;147(12):1133-1139. doi:10.1093/oxfordjournals.aje.a009411
- Koschinsky ML, Bajaj A, Boffa MB, et al. A focused update to the 2019 NLA scientific statement on use of lipoprotein(a) in clinical practice. J Clin Lipidol. 2024;18(3):e308-e319. doi:10.1016/j.jacl.2024.03.001