
Is Heart Disease Optional? The New Science that Could End Heart Attacks Forever
The Hook: Is Getting Older a Death Sentence for Your Heart?
Most people think that “clogged arteries” are just a normal part of getting old. We treat heart disease like grey hair or wrinkles—something that eventually happens to everyone if they live long enough. But what if that is wrong?
New research is pointing toward something called the Zero-Risk Hypothesis. This idea suggests that heart disease isn’t a natural part of aging. Instead, it is a problem caused by having too much “stuff” in our blood for too long. If we keep that “stuff” very low from birth, heart disease might actually be optional. Let’s look at the surprising findings that show how we could effectively erase the world’s biggest killer.
Takeaway 1: It’s Not Aging; It’s the “Trap” in Your Arteries
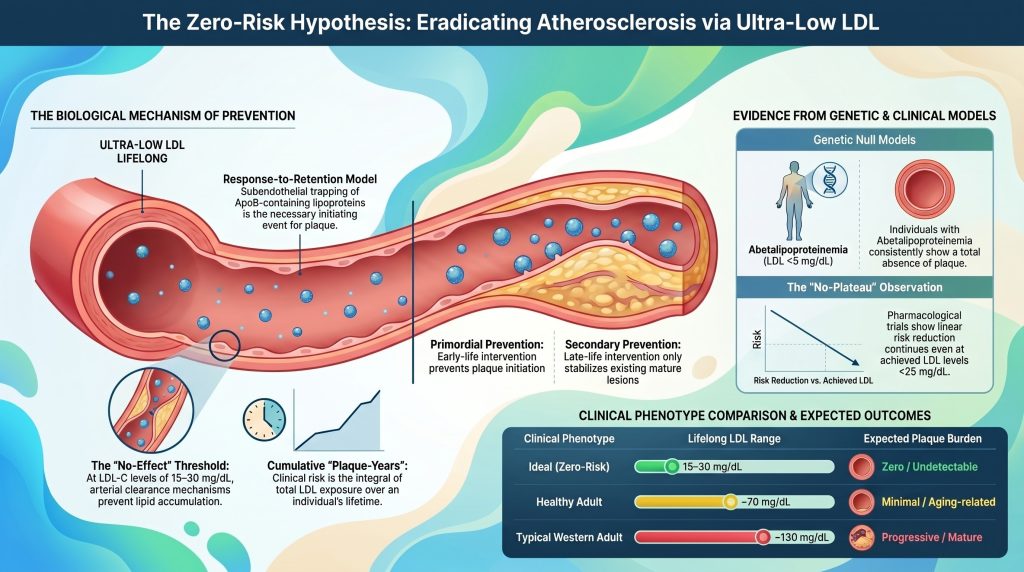
To understand how heart disease starts, scientists use the Response-to-Retention model. Think of your artery wall like a sticky trap on the floor of a house. In your blood, there are tiny particles called ApoB. These particles are like little bugs crawling around.
Every single “bad” cholesterol particle contains exactly one molecule of ApoB. This makes ApoB the perfect way to count how many “bugs” are in your system. Heart disease—or atherosclerosis—starts when these particles get stuck in the “trap” under the lining of your artery wall. Once they are trapped, they rot and cause a mess. This creates a buildup called plaque.
The big secret? If there are no particles to get trapped, the disease simply cannot start. It doesn’t matter how old you are. If the “trap” stays empty, your arteries stay clean. This changes our view of heart disease. It isn’t about “bad luck.” It is about biological chemistry. If we control the chemistry, we take luck out of the equation.
Takeaway 2: The Concept of “Plaque-Years”
Think of heart risk like a ticking clock. Doctors call this “cumulative exposure.” The more particles you have in your blood, and the longer they stay there, the faster your clock ticks toward a heart attack. Scientists call this measurement plaque-years.
You can actually calculate your own risk with simple math. A plaque-year is roughly your LDL cholesterol level multiplied by your age. If your LDL is high, your “timer” runs fast. If it is very low, the timer barely moves. For men, researchers have found specific levels where the risk of a heart event becomes much higher.
| Risk Level | Plaque-Years (LDL x Age) | Chance of a Major Heart Event |
| Low Risk | Less than 5,000 | Less than 1% chance. |
| Intermediate | Around 8,000 | Average risk for your age. |
| High Risk | Around 12,500 | 5% probability of an event. |
| Very High Risk | Over 16,000 | Treated as if disease is already there. |
By keeping these levels ultra-low from a young age, you can stay in the “Low Risk” zone forever. As the research states:
“under the zero-risk hypothesis, maintaining an LDL-C of 15 mg/dL for an 80-year lifespan results in exposure that is vastly below the threshold for even minimal risk.”
Takeaway 3: Nature’s “Zero-Plaque” Experiments
How do we know for sure that heart disease can be stopped? We look at “experiments of nature.” Some people are born with rare genes that keep their cholesterol ultra-low.
One group has a condition called Abetalipoproteinemia. These people have almost zero heart disease particles. When doctors study them, they find their arteries are completely free of plaque, even when they get old. This proves that without the particles, the disease does not exist.
However, those people can sometimes be sickly. The real “superstars” of heart health are people with mutations in genes called PCSK9 or ANGPTL3. These people are perfectly healthy and fertile, but they have naturally ultra-low LDL. Scientists even found one healthy woman with an LDL level of only 15 mg/dL! These “healthy” versions prove we can have very low levels and still be strong, while our arteries stay as clean as a newborn’s.
Takeaway 4: “Lower is Better” Has No Limit
For a long time, people were worried about having cholesterol levels that were “too low.” They thought the body might need a certain amount to work right. However, huge medical studies (like the FOURIER and ODYSSEY trials) have shown that there is no limit to the benefits.
When patients dropped their levels as low as 15 mg/dL, they continued to see fewer heart problems. More importantly, they stayed safe. The biology suggests our bodies handle very low levels perfectly fine. In fact, our hearts actually prefer it. It’s like a car engine; it runs much longer if the fuel is perfectly clean.
Takeaway 5: Turning “Vulnerable Pimples” into “Solid Warts”
What if you already have some buildup in your arteries? Ultra-low levels can still help by changing what the plaque is made of.
Dangerous plaque is like a vulnerable pimple. It is soft, full of fat, and can pop easily. If it pops, it causes a heart attack. But when you lower LDL to extreme levels, the body pulls the fat out of that “pimple.” It replaces the fat with tough tissue called collagen.
This process “cements” the plaque. As the research notes, it turns a “vulnerable pimple” into a “solid wart.” A solid wart is stable. It is much less likely to pop and cause a sudden heart attack.
Takeaway 6: Inflammation Needs a Partner
You may have heard that “inflammation” causes heart disease. This is true, but it’s only half the story. Inflammation is like a fire, but the heart disease particles (LDL) are the fuel.
You can have a lot of inflammation in your body from things like stress or being sick. But if there is no “fuel” (no trapped particles) in your artery walls, a fire cannot start there. Inflammation is a partner to heart disease, but it cannot cause the disease all by itself. If you remove the fuel, you stop the fire. Even if the “match” of inflammation is lit, there is nothing in the artery wall to burn.
Closing: The Future of “Primordial” Prevention
We are entering a new era of medicine called primordial prevention. Most doctors today try to fix a heart that is already damaged. Primordial prevention is different. The goal is to never let the damage start in the first place. This means keeping “heart disease particles” low from the time we are born.
If we can do this, we might look back on heart disease the same way we look at polio or smallpox. We will see it as a scary problem from the past that we eventually learned how to erase.
The Final Thought: If we have the tools to stop the “trap” from ever catching a single particle, could heart disease become a choice rather than a fate? Future generations may wonder why we ever let it happen in the first place.
DEEP DIVE
Is Atherosclerosis Biologically Eliminated at Ultra-Low Lifelong LDL Levels?
A Critical Evaluation of the “Zero-Risk” Hypothesis
Atherosclerotic cardiovascular disease (ASCVD) has long been framed as an inevitable consequence of human senescence, yet contemporary molecular biology and genetic epidemiology increasingly converge on a substrate-dependent model in which the disease is, in principle, biologically avoidable [1], [2]. The premise of the “zero-risk” hypothesis is that atherosclerosis is primarily an apolipoprotein B (ApoB)–driven disorder [2], [3]. By maintaining lifelong exposure to ultra-low levels of low-density lipoprotein cholesterol (LDL-C) and its corresponding ApoB particle concentration—approximately 10–20 mg/dL (0.26–0.52 mmol/L), near the physiological newborn baseline—the probability of initiating the pathognomonic lesion of atherosclerosis (subendothelial retention of atherogenic lipoproteins) may be reduced to a level that is biologically negligible [2], [4]. This report evaluates whether such ultra-low exposure effectively eliminates disease initiation, or whether residual pathways involving inflammation, lipoprotein(a) [Lp(a)], and endothelial dysfunction retain the capacity to seed arterial plaque independently of ApoB [5], [6].

The Response-to-Retention Model: A Deterministic Framework for Initiation
The theoretical basis for the biological elimination of atherosclerosis rests on the response-to-retention hypothesis, which identifies subendothelial trapping of ApoB-containing lipoproteins as the necessary and sufficient initiating event for atherogenesis [1], [7]. While traditional models emphasized frank endothelial injury as the primary trigger, current evidence indicates that an intact, though dysfunctional, endothelium typically overlies early and intermediate lesions [1], [8]. Disease initiation occurs when lipoproteins smaller than approximately 70 nm in diameter—including LDL, triglyceride-rich remnants, and Lp(a)—traverse the endothelial barrier and enter the tunica intima [2], [7].
Molecular Interactions within the Intimal Matrix
Within the intima, retained particles interact with the extracellular matrix (ECM), particularly with negatively charged proteoglycans such as versican, perlecan, biglycan, and decorin [1], [7]. Positively charged regions of ApoB, specifically sequences rich in lysine and arginine residues, bind electrostatically to the glycosaminoglycan (GAG) chains of these proteoglycans [1]. This is not passive entrapment; it is a critical biochemical event that prolongs the residence time of the lipoprotein within the arterial wall by an order of magnitude relative to plasma transit [9].
| Proteoglycan | Interaction with ApoB Lipoproteins | Role in Atherogenesis |
| Versican | Large aggregating proteoglycan that expands intimal volume | Promotes lipoprotein trapping and smooth muscle cell migration [7] |
| Perlecan | Basement-membrane heparan sulfate proteoglycan | Structural scaffold facilitating early particle retention [7] |
| Biglycan | Small leucine-rich proteoglycan with high ApoB affinity | Correlates with development of lipid-rich necrotic core [7] |
| Decorin | Interacts with collagen and LDL particles | Modulates fibrotic response and lipoprotein aggregation [7] |
Prolonged intimal residence renders retained lipoproteins highly susceptible to chemical modification, including oxidation, glycation, enzymatic cleavage, and aggregation [1], [9]. Modified particles trigger a maladaptive immune response [1], [7]. Endothelial cells activated by modified lipids and mechanical stressors express adhesion molecules (VCAM-1, ICAM-1) and secrete chemokines that recruit monocytes into the subendothelial space [1], [10]. These monocytes differentiate into macrophages that internalize modified lipoproteins via scavenger receptors and transform into lipid-laden foam cells—the morphological hallmark of the fatty streak [10].
Thermodynamic and Kinetic Limits of Retention
The probability of particle retention is a function of the local concentration of ApoB particles and the availability of proteoglycan binding sites within the matrix [7]. In a stochastic model of the arterial wall, the rate of lipid accumulation is determined by the flux of lipoproteins into the intima and the kinetics of proteoglycan binding [9]. At ultra-low circulating concentrations (e.g., LDL-C ≈ 15 mg/dL), the number of particles available for transcytosis is vastly reduced [2], [11].
If the rate of particle entry is low enough that the arterial wall’s endogenous clearance mechanisms—macrophage-mediated phagocytosis and HDL-mediated reverse cholesterol transport—can remove particles before pro-inflammatory modification, the inflammatory cascade is never initiated [1], [12]. This is the mechanistic basis for a biological “no-effect” threshold for ApoB concentration, below which the probability of initiating a lesion approaches zero [7].
The Quantitative Relationship of Cumulative Exposure: Plaque-Years
Clinical risk of ASCVD reflects not instantaneous LDL-C but the integral of exposure over time, a concept now formalized as “cumulative LDL exposure” or “plaque-years” [13], [14]. Under this framework, atherosclerosis is a disease of gradual substrate accumulation, and the time to a clinical event is determined by how quickly an individual crosses a personal plaque threshold [13]. Domanski and colleagues formally demonstrated in a pooled analysis of 4,958 participants from ARIC, CARDIA, MESA, and the Framingham Offspring Study that the cumulative burden of LDL-C exposure, the time course of that exposure, and the slope of LDL-C change over time each independently predicted incident cardiovascular events [14].
Defining the Plaque-Year Thresholds
Analysis of epidemiologic and Mendelian randomization data permits tentative quantification of cumulative-exposure thresholds [13], [15]. Published thresholds remain approximate and cohort-dependent rather than formally validated across populations, and the values below are best regarded as heuristic estimates drawn from long-term cohort and Mendelian randomization data [13]–[15].
| Risk Metric (Men) | Approx. Cumulative LDL Exposure (g·yr/dL) | Lifetime Major Event Risk (approx.) |
| Low-Risk Threshold | ~ 5,000 | < 10% probability of event [13], [14] |
| Intermediate Threshold | ~ 8,000 | Approximate median age for non-zero CAC (~age 55–60) [13], [16] |
| High-Risk Threshold | ~ 11,000 | >20% probability of event [13], [14] |
| Very High-Risk Threshold | ~ 14,000 | Approximate median age for CAC ≥ 100 [13], [16] |
Note: Thresholds for women are generally higher (estimated by roughly 20–30% at comparable risk strata), consistent with the observed later-life onset of ASCVD in women and premenopausal attenuation of LDL transcytosis and arterial-wall biology [13].
Influence of Secondary Risk Factors on the Retention Threshold
Cumulative LDL exposure required to initiate events is not static; it is modulated by the biological environment of the artery [13]. Hypertension, diabetes, and smoking effectively lower the threshold by increasing particle retention or accelerating inflammatory response to retained lipids [2], [17].
- Hypertension: Elevated blood pressure increases LDL transcytosis across the endothelium and promotes synthesis of proteoglycans with higher ApoB affinity [7]. Shear stress on existing plaques raises rupture likelihood [13].
- Type 2 diabetes / insulin resistance: Chronic dysglycemia and hyperinsulinemia injure the arterial wall, promote adverse remodeling, and narrow the coronary lumen, so even small plaques become hemodynamically significant and thrombi more likely to be occlusive [14].
- Smoking: Tobacco exposure causes direct endothelial damage and oxidative stress, accelerating intimal lipoprotein modification [2].
- Androgen abuse: In young male anabolic-androgenic steroid users, plaque volume and coronary artery calcium (CAC) scores correlate strongly with lifetime exposure, consistent with an independent acceleration of atherogenesis [18].
For a metabolically healthy individual with controlled blood pressure and no inflammatory comorbidity, the arterial wall can tolerate a higher cumulative burden before clinical disease manifests [13]. Under the zero-risk hypothesis, maintaining LDL-C ≈ 15 mg/dL across an 80-year lifespan yields only ~1,200 g·yr/dL of cumulative exposure—well below the ~5,000 g·yr/dL heuristic low-risk threshold and roughly an order of magnitude below thresholds associated with clinically meaningful event probabilities [13].
Genetic Null Models: Experiments of Nature
The most rigorous test of whether atherosclerosis can be eliminated lies in human genetic models of lifelong ultra-low ApoB exposure [2], [3]. These “experiments of nature” provide the strongest available evidence that, in the near-absence of atherogenic particles, disease initiation is profoundly attenuated [19], [20]. Case-report numbers remain small and autopsy data are limited, so claims of “complete” absence should be interpreted as “markedly reduced burden beyond what is plausibly attributable to chance alone” rather than mathematically zero.
Abetalipoproteinemia (ABL) and MTTP Deficiency
Abetalipoproteinemia is an autosomal recessive disorder caused by biallelic loss-of-function mutations in MTTP, which is essential for assembly and secretion of ApoB-containing lipoproteins in the liver and intestine [19], [21]. Affected individuals have near-total absence of ApoB-containing lipoproteins in circulation; total cholesterol is typically < 30 mg/dL and LDL-C < 5 mg/dL (often undetectable) [19].
Despite severe non-cardiac pathology (fat malabsorption, acanthocytosis, spinocerebellar degeneration, retinitis pigmentosa), clinical and limited autopsy evidence indicate a striking paucity of atherosclerotic lesions in ABL patients [19], [21]. Cardiomyopathy and arrhythmias, when they occur, are attributed predominantly to fat-soluble vitamin deficiency (vitamin E) rather than ischemia [19]. ABL thus functions as the closest available human null model for ApoB-driven atherosclerosis.
Familial Hypobetalipoproteinemia (FHBL)
FHBL results from heterozygous or biallelic APOB mutations producing truncated, secretion-incompetent proteins [22].
- Heterozygous FHBL: LDL-C typically 20–50 mg/dL, with markedly reduced lifetime ASCVD risk [22], [23].
- Homozygous / compound heterozygous FHBL: LDL-C often < 10 mg/dL; phenotype resembles ABL and exhibits similarly marked protection against atherosclerotic disease on imaging and in available autopsy reports [22], [23].
| Genetic Disorder | Mechanism | LDL-C (mg/dL) | ASCVD Phenotype |
| Abetalipoproteinemia | MTTP LOF; no particle assembly | < 5 | Markedly reduced / absent plaque [19] |
| Homozygous FHBL | APOB LOF; truncated proteins | < 10 | Markedly reduced / absent plaque [22], [23] |
| PCSK9 LOF (compound het.) | Enhanced LDLR recycling; rapid LDL clearance | ~ 14–15 | Healthy phenotype; no documented ASCVD in index case [24] |
| ANGPTL3 Deficiency | Increased LPL/EL activity; low TG and LDL | ~ 30–40 | Markedly reduced CAD (~34–41% lower odds) [25], [26] |
PCSK9 and ANGPTL3: The “Healthy” Ultra-Low Phenotype
Unlike ABL, which carries severe non-cardiac morbidity, loss-of-function variants in PCSK9 and ANGPTL3 produce ultra-low LDL-C without fat malabsorption or hepatic steatosis [3], [27]. The first reported compound heterozygous PCSK9 LOF individual, a healthy fertile African American woman described by Zhao and colleagues, had an LDL-C of approximately 14 mg/dL (0.36 mmol/L) and was clinically unremarkable, demonstrating that lifelong near-absence of circulating PCSK9 is compatible with normal human physiology [24]. In the Atherosclerosis Risk in Communities (ARIC) cohort, heterozygous PCSK9 LOF variants (PCSK9 Y142X and C679X) were associated with a 28% lower LDL-C and an 88% reduction in coronary heart disease incidence over approximately 15 years in Black participants, with a more modest ~47% reduction tied to PCSK9 R46L carriers in White participants [28]. Extended over a lifetime of exposure, the risk reduction approaches the near-complete protection observed in ABL and homozygous FHBL [2].
ANGPTL3 deficiency produces “combined hypolipidemia” with low LDL-C, HDL-C, and triglycerides [27]. In Stitziel’s 2017 analysis, three compound-heterozygous individuals had zero coronary plaque on CT angiography versus a mean 39% plaque burden in matched relatives; heterozygous LOF carriers exhibited approximately 34% lower odds of coronary artery disease (OR 0.66; 95% CI 0.44–0.98) [25]. Dewey and colleagues replicated this signal in 58,335 DiscovEHR participants, reporting a 41% lower odds of CAD (OR 0.59; 95% CI 0.41–0.85; p = 0.004) [26]. Critically, quantitative imaging confirms that ANGPTL3 LOF carriers do not have increased hepatic fat, distinguishing this pathway as a favorable target for long-term pharmacological mimicry [27].
Pharmacologic Evidence: The “Lower Is Better” Paradigm
Genetic models speak to lifelong exposure; pharmacologic trials test the effect of lowering LDL-C later in life, typically in individuals with existing subclinical or clinical disease [2].
Meta-Regression and the Linearity of Benefit
Data from the Cholesterol Treatment Trialists (CTT) Collaboration and subsequent non-statin trials (ezetimibe, PCSK9 inhibitors) demonstrate a remarkably consistent dose-response: every 1 mmol/L (38.7 mg/dL) reduction in LDL-C corresponds to approximately a 22% reduction in major vascular events per year of treatment, with the relationship remaining linear even at the lowest achieved levels examined [29], [30]. This relationship is effectively agnostic to the mechanism by which LDL-C is lowered [15], [31].
| Clinical Trial / Analysis | Achieved LDL-C (mg/dL) | Key Finding |
| CTT Meta-analysis | Range 60–180 | ~22% RR reduction per 1 mmol/L (38.7 mg/dL) [29] |
| IMPROVE-IT (ezetimibe + simvastatin) | 53.7 vs 69.5 (TWA) | HR 0.936 (0.89–0.99) for primary endpoint; benefit beyond statin alone [30] |
| FOURIER (evolocumab) | Median ~30 (subgroup <20) | Linear benefit continued to LDL-C < 20 mg/dL [32], [33] |
| ODYSSEY OUTCOMES (alirocumab) | ~53 (48-wk mean) | HR 0.85 MACE; first mortality signal for PCSK9i (HR 0.85) [34] |
| PROLONG-ANG3 (solbinsiran) | Phase 2; sustained reduction | siRNA targeting ANGPTL3; durable ApoB and TG lowering [35] |
| CORALreef Lipids (enlicitide) | ~60 on background statin | Oral PCSK9 inhibitor; Phase 3 LDL-C reduction [36] |
The significance of these pharmacologic data lies in the “no-plateau” observation [32], [37]. Event reduction continues linearly even at achieved LDL-C below 20 mg/dL, suggesting that the biological drivers of atherosclerosis remain substrate-limited at the extremes of the lipid spectrum [2], [32].
Residual Disease versus New Initiation
A critical distinction must be drawn between primary and secondary prevention [13]. Among trial participants achieving very low LDL-C, a meaningful fraction still experience events [38]. This residual risk is not a failure of the zero-risk hypothesis but rather the predictable consequence of the irreversible structural features of advanced plaque [2]. Once a lesion has developed a necrotic core and a thinned fibrous cap, rupture can be triggered by local mechanical forces or systemic inflammation largely independent of current LDL-C [2], [39]. The zero-risk hypothesis concerns disease initiation, not terminal rupture of pre-existing plaque [1]. Were ultra-low lipoprotein levels maintained from birth, the substrate for acute events (mature, unstable plaque) would simply fail to form [2], [14].
Residual Risk Pathways: Can Inflammation or Lp(a) Initiate Disease Alone?
Falsifying the zero-risk hypothesis requires identifying a non-ApoB pathway capable of independently initiating atherosclerosis in the absence of atherogenic lipoproteins [5], [6].
Lipoprotein(a) and the Structural Necessity of ApoB
Lp(a) is a complex particle consisting of an LDL-like ApoB-100 core covalently bound to apolipoprotein(a) [40]. It is independently associated with ASCVD risk even when LDL-C is optimally controlled [5]. Mechanistically, Lp(a) is more atherogenic than LDL on a per-particle basis because of its propensity for matrix binding and its cargo of oxidized phospholipids (OxPLs), which drive robust inflammation [40], [41]. Critically, however, every Lp(a) particle requires an ApoB-100 scaffold; in ABL and homozygous FHBL, Lp(a) cannot be synthesized because ApoB secretion is absent [19], [22]. Thus, Lp(a) does not represent a non-ApoB initiation pathway but a quantitatively more atherogenic subclass of the ApoB-containing lipoprotein family.
Inflammation as a Potentiator, Not an Initiator
The role of inflammation (IL-6, IL-1β, hsCRP) in ASCVD is well established, and the CANTOS trial demonstrated that canakinumab-mediated blockade of IL-1β reduces cardiovascular events without changing lipid levels [42]. Nevertheless, current consensus is that inflammation operates as a physiologic response of the arterial wall to retained lipoproteins rather than as an autonomous initiator [7]. In wild-type animal models, systemic inflammation does not generate atherosclerosis in the absence of hyperlipidemia [43]. Arterial-wall inflammation, monocyte recruitment, and foam-cell formation are downstream consequences of lipoprotein retention [1]. Chronic inflammatory diseases (e.g., rheumatoid arthritis, SLE) lower the threshold for retention-driven lesion formation, but they do not initiate atherosclerosis where the ApoB substrate is absent [7].
Imaging the Ultra-Low End: Subclinical Evidence
Coronary artery calcium (CAC) scoring, intravascular ultrasound (IVUS), optical coherence tomography (OCT), and coronary CT angiography (CCTA) provide complementary windows into the presence and composition of subclinical plaque at extreme lipid strata [16], [44].
The Power of Zero CAC
A CAC score of zero is a robust marker of low short-term event risk, but its interpretation is age-dependent [16], [45]. In younger populations (age < 45), zero CAC is common even among those with significant LDL exposure because calcification is a late-stage stabilization marker [45]. Conversely, zero CAC in an elderly individual is uncommon and identifies exceptional resistance to atherogenesis, often associated with genetically low ApoB or favorable matrix biology [16], [46].
| CAC Category | Agatston Units | Clinical Significance |
| Zero | 0 | No detectable calcification; very low short-term ASCVD risk [16] |
| Mild | 1–99 | Early atherosclerotic burden; supports risk up-classification [16] |
| Moderate | 100–399 | Significant plaque; 10-yr event risk typically exceeds 7.5% [16] |
| Severe | ≥ 400 | Very high risk; often treated as ASCVD equivalent [16] |
IVUS and the Threshold for Regression
Intravascular imaging trials consistently show that deep LDL-C lowering can reverse net plaque burden [47], [48]. The ASTEROID trial achieved mean LDL-C of 60.8 mg/dL with rosuvastatin 40 mg and demonstrated regression of percent atheroma volume (PAV) across multiple imaging parameters [47]. SATURN corroborated regression at LDL-C in the 60–70 mg/dL range [48]. GLAGOV extended the relationship into the PCSK9-inhibitor era, showing continuous linear regression with evolocumab down to LDL-C < 40 mg/dL, with no apparent plateau [49].
- Regression: Reduction in PAV is observed consistently when LDL-C falls below ~70 mg/dL, with the magnitude of regression scaling with the depth of LDL-C reduction [47]–[49].
- Stabilization: At ultra-low levels, plaques undergo beneficial remodeling; the lipid-rich core shrinks and the fibrous cap thickens and becomes collagen-rich, converting a “vulnerable” lesion into a structurally stable one [49], [50].
Under the zero-risk hypothesis, early-life maintenance of LDL-C ≈ 15 mg/dL would prevent formation of the lipid-rich core entirely; regression and stabilization become moot because there is no lesion to regress [2].
Temporal Considerations: Early-Life Exposure versus Adult Intervention
The cumulative-exposure framework emphasizes that the age at which LDL lowering begins matters as much as its intensity [13].
The Window of Opportunity
Ference and colleagues’ landmark 2012 Mendelian randomization analysis pooled nine SNPs across six LDL-regulating genes and found that each standard unit of genetically lower LDL-C produced approximately a three-fold greater proportional reduction in coronary heart disease than the same magnitude of late-life statin-induced LDL-C lowering [15]. Specifically, lifelong exposure to ~1 mmol/L lower LDL-C was associated with roughly a 54–55% reduction in CHD risk, versus approximately a 22% relative risk reduction per mmol/L with statin therapy initiated in midlife [15], [29]. This discrepancy arises because early intervention prevents lesion initiation, whereas late intervention merely slows progression of already-mature plaques [2], [14].
For a metabolically healthy individual:
- Maintaining LDL-C ≈ 15 mg/dL lifelong: Prevents plaque initiation; lifetime risk approaches zero [2], [15].
- Reducing LDL-C to ≈ 15 mg/dL at age 40: Halts further progression, stabilizes existing subclinical plaque, and delays clinical events by an estimated 10–20 years depending on baseline plaque burden [13], [14].
- Reducing LDL-C to ≈ 15 mg/dL after a clinical event: Reduces recurrence risk by stabilizing vulnerable plaques; does not eliminate residual risk arising from irreversible structural damage [34], [50].
The biological primacy of primary—and ideally primordial—prevention follows directly: maintaining physiologic lipoprotein levels from the earliest stages of life avoids the transition from subclinical to clinically dangerous burden [3].
Theoretical and Mechanistic Limits: Stochastic versus Zero Risk
Is the elimination of atherosclerosis absolute, or does a biological floor persist?
Stochastic Retention at Extreme Lows
Atherosclerosis initiation is fundamentally a probabilistic process [7]. Even at LDL-C ≈ 15 mg/dL, an ApoB particle could in principle enter the arterial wall, bind a proteoglycan, and undergo oxidation [7]. However, the probability of enough such particles being retained in a single focal region to trigger a self-perpetuating inflammatory response becomes vanishingly small at ultra-low concentrations [7], [9].
Arterial-wall biology incorporates multiple fail-safe mechanisms:
- Resident scavenging: Healthy intimal macrophages can clear small quantities of modified lipoprotein without becoming foam cells or secreting pro-inflammatory cytokines [1].
- HDL/ApoA-I efflux: HDL functions as a bidirectional lipid vector capable of removing excess cholesterol from the intima; at low ApoB influx, efflux capacity easily maintains homeostatic balance [8], [51].
- Matrix integrity: In the absence of sustained lipid retention, the intima remains thin and structurally intact, with preserved elastic fibers and minimal proteoglycan expansion [9].
At the zero-risk target of LDL-C ≈ 15 mg/dL, the probability of plaque initiation may not be mathematically zero but is effectively zero in biological and clinical terms: accumulation would never reach the critical mass required for disease within a human lifespan [2], [14]. The Horus study of 137 mummies across 4,000 years of history—including Tsimane-like pre-industrial and ancient peoples—documents vascular calcification in every era examined, but notably, pre-industrial populations with very low LDL-C (e.g., the contemporary Tsimane, with mean LDL-C ~91 mg/dL and the lowest prevalence of coronary atherosclerosis measured in any human population) exhibit only low rates of subclinical CAC even in the elderly [52], [53]. Extrapolating to lifelong LDL-C of 15 mg/dL implies burden far below even this baseline.
Synthesis of the Zero-Risk Hypothesis Outcomes
Three hypothesis variants can be tested against the evidence reviewed above.
Strong Form: Zero-Risk Hypothesis
Lifelong LDL-C ≈ 15 mg/dL in a metabolically healthy individual produces effectively zero probability of clinically meaningful atherosclerosis. Evaluation: supported by human genetic null models (ABL, homozygous FHBL) where atherosclerotic burden is markedly reduced or absent [19], [22], by the physiologic newborn LDL-C baseline, and by the consistent absence of events in ultra-low-exposure genetic cohorts [2], [24].
Weak Form: Asymptotic Hypothesis
Risk approaches zero but never fully reaches it because of stochastic retention or non-ApoB pathways. Evaluation: supported by the fundamentally probabilistic nature of particle–wall interactions [7]. Extreme age or severe systemic inflammation could, in principle, cause minimal intimal changes, though such lesions would likely remain subclinical across the human lifespan [7].
Null Hypothesis
Atherosclerosis still occurs at meaningful rates independent of LDL at very low levels. Evaluation: refuted for the ideal phenotype [2], [15]. Residual risk observed in treated secondary-prevention populations is consistently attributable to pre-existing plaque and to non-LDL ApoB-containing particles (triglyceride-rich remnants and Lp(a)) rather than to a non-ApoB mechanism of initiation [5], [40].
| Clinical Phenotype | Expected Plaque Burden | Event Risk Probability |
| Ideal phenotype (lifelong LDL-C ~15 mg/dL) | Zero / undetectable | Effectively zero [15] |
| Healthy adult (lifelong LDL-C ~70 mg/dL) | Minimal / aging-related | Low (~5–10%) [14] |
| Western adult (lifelong LDL-C ~130 mg/dL) | Progressive / mature | High [2], [14] |
| Secondary prevention (achieved LDL-C ~15 mg/dL) | Stable / calcified | Significant residual [34] |
Conclusion: Redefining the Threshold of Human Health
The evaluation of the zero-risk hypothesis supports the view that atherosclerosis is a substrate-dependent disease requiring a minimum lifelong cumulative exposure to ApoB-containing lipoproteins to initiate [2], [14]. The quantitative relationship between cumulative exposure and plaque formation implies that maintaining LDL-C in the 10–20 mg/dL range from birth delays disease initiation beyond the biological limits of human longevity [13], [15].
Genetic models (abetalipoproteinemia, homozygous FHBL, and healthy ultra-low phenotypes from PCSK9 and ANGPTL3 loss of function) demonstrate that such low levels are both safe and compatible with a marked reduction—and in some cases apparent absence—of atherosclerotic plaque, acknowledging that case numbers remain limited [19], [22], [24], [25]. While residual risks driven by Lp(a), remnants, and inflammation persist in patients with mature disease, these factors are insufficient to initiate atherogenesis in the near-total absence of ApoB particles [2], [40].
The implication for preventive cardiology is substantial [3]. Rather than managing risk as an inevitable consequence of aging, the proper objective is primordial prevention: maintaining physiologic lipoprotein levels throughout the earliest stages of life to eliminate the substrate for atherogenesis [2], [15]. This paradigm suggests that heart disease—the leading cause of death worldwide—is biologically eradicable if ApoB particle retention is prevented throughout the human lifespan [2], [14].
References
- J. Borén, M. J. Chapman, R. M. Krauss, C. J. Packard, J. F. Bentzon, C. J. Binder, M. J. Daemen, L. L. Demer, R. A. Hegele, S. J. Nicholls, B. G. Nordestgaard, G. F. Watts, E. Bruckert, S. Fazio, B. A. Ference, I. Graham, J. D. Horton, U. Landmesser, U. Laufs, L. Masana, G. Pasterkamp, F. J. Raal, K. K. Ray, H. Schunkert, M.-R. Taskinen, B. van de Sluis, O. Wiklund, L. Tokgözoğlu, A. L. Catapano, and H. N. Ginsberg, “Low-density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights—a consensus statement from the European Atherosclerosis Society Consensus Panel,” Eur. Heart J., vol. 41, no. 24, pp. 2313–2330, Jun. 2020, doi: 10.1093/eurheartj/ehz962.
- B. A. Ference, H. N. Ginsberg, I. Graham, K. K. Ray, C. J. Packard, E. Bruckert, R. A. Hegele, R. M. Krauss, F. J. Raal, H. Schunkert, G. F. Watts, J. Borén, S. Fazio, J. D. Horton, L. Masana, S. J. Nicholls, B. G. Nordestgaard, B. van de Sluis, M.-R. Taskinen, L. Tokgözoğlu, U. Landmesser, U. Laufs, O. Wiklund, J. K. Stock, M. J. Chapman, and A. L. Catapano, “Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies,” Eur. Heart J., vol. 38, no. 32, pp. 2459–2472, Aug. 2017, doi: 10.1093/eurheartj/ehx144.
- A. L. Catapano, I. Graham, G. De Backer, O. Wiklund, M. J. Chapman, H. Drexel, A. W. Hoes, C. S. Jennings, U. Landmesser, T. R. Pedersen, Ž. Reiner, G. Riccardi, M.-R. Taskinen, L. Tokgözoğlu, W. M. M. Verschuren, P. Vlachopoulos, D. A. Wood, and J. L. Zamorano, “2016 ESC/EAS guidelines for the management of dyslipidaemias,” Eur. Heart J., vol. 37, no. 39, pp. 2999–3058, Oct. 2016, doi: 10.1093/eurheartj/ehw272.
- J. H. O’Keefe, L. Cordain, W. H. Harris, R. M. Moe, and R. Vogel, “Optimal low-density lipoprotein is 50 to 70 mg/dl: lower is better and physiologically normal,” J. Am. Coll. Cardiol., vol. 43, no. 11, pp. 2142–2146, Jun. 2004, doi: 10.1016/j.jacc.2004.03.046.
- B. G. Nordestgaard, M. J. Chapman, K. Ray, J. Borén, F. Andreotti, G. F. Watts, H. Ginsberg, P. Amarenco, A. Catapano, O. S. Descamps, E. Fisher, P. T. Kovanen, J. A. Kuivenhoven, P. Lesnik, L. Masana, Z. Reiner, M. R. Taskinen, L. Tokgözoğlu, and A. Tybjærg-Hansen, “Lipoprotein(a) as a cardiovascular risk factor: current status,” Eur. Heart J., vol. 31, no. 23, pp. 2844–2853, Dec. 2010, doi: 10.1093/eurheartj/ehq386.
- B. G. Nordestgaard and A. Varbo, “Triglycerides and cardiovascular disease,” Lancet, vol. 384, no. 9943, pp. 626–635, Aug. 2014, doi: 10.1016/S0140-6736(14)61177-6.
- I. Tabas, K. J. Williams, and J. Borén, “Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications,” Circulation, vol. 116, no. 16, pp. 1832–1844, Oct. 2007, doi: 10.1161/CIRCULATIONAHA.106.676890.
- P. Libby, “The changing landscape of atherosclerosis,” Nature, vol. 592, no. 7855, pp. 524–533, Apr. 2021, doi: 10.1038/s41586-021-03392-8.
- D. C. Schwenke and T. E. Carew, “Initiation of atherosclerotic lesions in cholesterol-fed rabbits. II. Selective retention of LDL vs. selective increases in LDL permeability in susceptible sites of arteries,” Arteriosclerosis, vol. 9, no. 6, pp. 908–918, Nov./Dec. 1989, doi: 10.1161/01.atv.9.6.908.
- R. Ross, “Atherosclerosis—an inflammatory disease,” N. Engl. J. Med., vol. 340, no. 2, pp. 115–126, Jan. 1999, doi: 10.1056/NEJM199901143400207.
- F. Mach, C. Baigent, A. L. Catapano, K. C. Koskinas, M. Casula, L. Badimon, M. J. Chapman, G. G. De Backer, V. Delgado, B. A. Ference, I. M. Graham, A. Halliday, U. Landmesser, B. Mihaylova, T. R. Pedersen, G. Riccardi, D. J. Richter, M. S. Sabatine, M.-R. Taskinen, L. Tokgözoğlu, and O. Wiklund, “2019 ESC/EAS guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk,” Eur. Heart J., vol. 41, no. 1, pp. 111–188, Jan. 2020, doi: 10.1093/eurheartj/ehz455.
- A. R. Tall and L. Yvan-Charvet, “Cholesterol, inflammation and innate immunity,” Nat. Rev. Immunol., vol. 15, no. 2, pp. 104–116, Feb. 2015, doi: 10.1038/nri3793.
- J. Brandts and K. K. Ray, “The LDL cumulative exposure hypothesis: evidence and practical applications,” Nat. Rev. Cardiol., vol. 21, no. 9, pp. 600–608, Sep. 2024, doi: 10.1038/s41569-024-01039-5.
- M. J. Domanski, X. Tian, C. O. Wu, J. P. Reis, A. K. Dey, Y. Gu, L. Zhao, S. Bae, K. Liu, A. A. Hasan, D. Zimrin, M. R. Farkouh, A. R. Hong, D. M. Lloyd-Jones, and V. Fuster, “Time course of LDL cholesterol exposure and cardiovascular disease event risk,” J. Am. Coll. Cardiol., vol. 76, no. 13, pp. 1507–1516, Sep. 2020, doi: 10.1016/j.jacc.2020.07.059.
- B. A. Ference, W. Yoo, I. Alesh, N. Mahajan, K. K. Mirowska, A. Mewada, J. Kahn, L. Afonso, K. A. Williams, and J. M. Flack, “Effect of long-term exposure to lower low-density lipoprotein cholesterol beginning early in life on the risk of coronary heart disease: a Mendelian randomization analysis,” J. Am. Coll. Cardiol., vol. 60, no. 25, pp. 2631–2639, Dec. 2012, doi: 10.1016/j.jacc.2012.09.017.
- R. Detrano, A. D. Guerci, J. J. Carr, D. E. Bild, G. Burke, A. R. Folsom, K. Liu, S. Shea, M. Szklo, D. A. Bluemke, D. H. O’Leary, R. Tracy, K. Watson, N. D. Wong, and R. A. Kronmal, “Coronary calcium as a predictor of coronary events in four racial or ethnic groups,” N. Engl. J. Med., vol. 358, no. 13, pp. 1336–1345, Mar. 2008, doi: 10.1056/NEJMoa072100.
- S. Yusuf, S. Hawken, S. Ôunpuu, T. Dans, A. Avezum, F. Lanas, M. McQueen, A. Budaj, P. Pais, J. Varigos, and L. Lisheng, “Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study,” Lancet, vol. 364, no. 9438, pp. 937–952, Sep. 2004, doi: 10.1016/S0140-6736(04)17018-9.
- A. L. Baggish, R. B. Weiner, G. Kanayama, J. I. Hudson, M. H. Picard, A. M. Hutter Jr., and H. G. Pope Jr., “Cardiovascular toxicity of illicit anabolic-androgenic steroid use,” Circulation, vol. 135, no. 21, pp. 1991–2002, May 2017, doi: 10.1161/CIRCULATIONAHA.116.026945.
- A. J. Hooper, B. A. Burnett, and J. R. Burnett, “Abetalipoproteinemia,” GeneReviews® [Internet], Univ. of Washington, Seattle, updated Jul. 2023. [Online]. Available: https://www.ncbi.nlm.nih.gov/books/NBK532447/.
- J. C. Cohen, E. Boerwinkle, T. H. Mosley Jr., and H. H. Hobbs, “Sequence variations in PCSK9, low LDL, and protection against coronary heart disease,” N. Engl. J. Med., vol. 354, no. 12, pp. 1264–1272, Mar. 2006, doi: 10.1056/NEJMoa054013.
- M. M. Berriot-Varoqueaux, L. P. Aggerbeck, M. Samson-Bouma, and J. R. Wetterau, “The role of the microsomal triglyceride transfer protein in abetalipoproteinemia,” Annu. Rev. Nutr., vol. 20, pp. 663–697, 2000, doi: 10.1146/annurev.nutr.20.1.663.
- S. G. Young and A. J. Hooper, “APOB-related familial hypobetalipoproteinemia,” GeneReviews® [Internet], Univ. of Washington, Seattle, updated Feb. 2021. [Online]. Available: https://www.ncbi.nlm.nih.gov/books/NBK570370/.
- L. Tarugi, E. Averna, E. Di Leo, A. Cefalù, D. Noto, S. Magnolo, M. Cattin, S. Bertolini, and M. Calandra, “Molecular diagnosis of hypobetalipoproteinemia: an ENID review,” Atherosclerosis, vol. 195, no. 2, pp. e19–e27, Dec. 2007, doi: 10.1016/j.atherosclerosis.2007.05.003.
- Z. Zhao, Y. Tuakli-Wosornu, T. A. Lagace, L. Kinch, N. V. Grishin, J. D. Horton, J. C. Cohen, and H. H. Hobbs, “Molecular characterization of loss-of-function mutations in PCSK9 and identification of a compound heterozygote,” Am. J. Hum. Genet., vol. 79, no. 3, pp. 514–523, Sep. 2006, doi: 10.1086/507488.
- N. O. Stitziel, A. V. Khera, X. Wang, A. J. Bierhals, A. C. Vourakis, A. E. Sperry, P. Natarajan, D. Klarin, C. A. Emdin, S. M. Zekavat, A. Nomura, J. Erdmann, H. Schunkert, N. J. Samani, W. E. Kraus, S. H. Shah, B. Yu, E. Boerwinkle, D. J. Rader, N. Gupta, P. M. Frossard, A. Rasheed, J. Danesh, E. S. Lander, S. Gabriel, D. Saleheen, K. Musunuru, and S. Kathiresan, “ANGPTL3 deficiency and protection against coronary artery disease,” J. Am. Coll. Cardiol., vol. 69, no. 16, pp. 2054–2063, Apr. 2017, doi: 10.1016/j.jacc.2017.02.030.
- F. E. Dewey, V. Gusarova, R. L. Dunbar, C. O’Dushlaine, C. Schurmann, O. Gottesman, S. McCarthy, C. V. Van Hout, S. Bruse, H. M. Dansky, J. B. Leader, M. F. Murray, M. D. Ritchie, H. L. Kirchner, D. H. Ledbetter, J. Penn, A. Lopez, I. B. Borecki, J. D. Overton, J. G. Reid, D. J. Carey, A. J. Murphy, G. D. Yancopoulos, A. Baras, J. Gromada, and A. R. Shuldiner, “Genetic and pharmacologic inactivation of ANGPTL3 and cardiovascular disease,” N. Engl. J. Med., vol. 377, no. 3, pp. 211–221, Jul. 2017, doi: 10.1056/NEJMoa1612790.
- K. Musunuru, J. P. Pirruccello, R. Do, G. M. Peloso, C. Guiducci, C. Sougnez, K. V. Garimella, S. Fisher, J. Abreu, A. J. Barry, T. Fennell, E. Banks, L. Ambrogio, K. Cibulskis, A. Kernytsky, E. Gonzalez, N. Rudzicz, J. C. Engert, M. A. DePristo, M. J. Daly, J. C. Cohen, H. H. Hobbs, D. Altshuler, G. Schonfeld, S. B. Gabriel, P. Yue, and S. Kathiresan, “Exome sequencing, ANGPTL3 mutations, and familial combined hypolipidemia,” N. Engl. J. Med., vol. 363, no. 23, pp. 2220–2227, Dec. 2010, doi: 10.1056/NEJMoa1002926.
- C. Baigent, L. Blackwell, J. Emberson, L. E. Holland, C. Reith, N. Bhala, R. Peto, E. H. Barnes, A. Keech, J. Simes, and R. Collins (Cholesterol Treatment Trialists’ Collaboration), “Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials,” Lancet, vol. 376, no. 9753, pp. 1670–1681, Nov. 2010, doi: 10.1016/S0140-6736(10)61350-5.
- Cholesterol Treatment Trialists’ (CTT) Collaboration, “Efficacy and safety of LDL-lowering therapy among men and women: meta-analysis of individual data from 174 000 participants in 27 randomised trials,” Lancet, vol. 385, no. 9976, pp. 1397–1405, Apr. 2015, doi: 10.1016/S0140-6736(14)61368-4.
- C. P. Cannon, M. A. Blazing, R. P. Giugliano, A. McCagg, J. A. White, P. Theroux, H. Darius, B. S. Lewis, T. O. Ophuis, J. W. Jukema, G. M. De Ferrari, W. Ruzyllo, P. De Lucca, K. Im, E. A. Bohula, C. Reist, S. D. Wiviott, A. M. Tershakovec, T. A. Musliner, E. Braunwald, and R. M. Califf, “Ezetimibe added to statin therapy after acute coronary syndromes,” N. Engl. J. Med., vol. 372, no. 25, pp. 2387–2397, Jun. 2015, doi: 10.1056/NEJMoa1410489.
- J. Silverman, L. A. Ballantyne, D. A. Bhatt, S. D. Wiviott, M. S. Sabatine, and Cholesterol Treatment Trialists, “Association between lowering LDL-C and cardiovascular risk reduction among different therapeutic interventions: a systematic review and meta-analysis,” JAMA, vol. 316, no. 12, pp. 1289–1297, Sep. 2016, doi: 10.1001/jama.2016.13985.
- M. S. Sabatine, R. P. Giugliano, A. C. Keech, N. Honarpour, S. D. Wiviott, S. A. Murphy, J. F. Kuder, H. Wang, T. Liu, S. M. Wasserman, P. S. Sever, and T. R. Pedersen, “Evolocumab and clinical outcomes in patients with cardiovascular disease,” N. Engl. J. Med., vol. 376, no. 18, pp. 1713–1722, May 2017, doi: 10.1056/NEJMoa1615664.
- R. P. Giugliano, T. R. Pedersen, A. C. Keech, A. Park, S. D. Wiviott, S. A. Murphy, J. F. Kuder, H. Wang, T. Liu, J. Lopez, and M. S. Sabatine, “Clinical efficacy and safety of achieving very low LDL-cholesterol concentrations with the PCSK9 inhibitor evolocumab: a prespecified secondary analysis of the FOURIER trial,” Lancet, vol. 390, no. 10106, pp. 1962–1971, Oct. 2017, doi: 10.1016/S0140-6736(17)32290-0.
- G. G. Schwartz, P. G. Steg, M. Szarek, D. L. Bhatt, V. A. Bittner, R. Diaz, J. M. Edelberg, S. G. Goodman, C. Hanotin, R. A. Harrington, J. W. Jukema, G. Lecorps, K. W. Mahaffey, A. Moryusef, R. Pordy, K. Quintero, M. T. Roe, W. J. Sasiela, J.-F. Tamby, P. Tricoci, H. D. White, A. M. Zeiher, and ODYSSEY OUTCOMES Committees and Investigators, “Alirocumab and cardiovascular outcomes after acute coronary syndrome,” N. Engl. J. Med., vol. 379, no. 22, pp. 2097–2107, Nov. 2018, doi: 10.1056/NEJMoa1801174.
- C. M. Ballantyne, J. P. Kastelein, L. A. Leiter, S. J. Nicholls, and colleagues, “Durability and efficacy of solbinsiran, a GalNAc-conjugated siRNA targeting ANGPTL3, in adults with mixed dyslipidaemia (PROLONG-ANG3): a double-blind, randomised, placebo-controlled, phase 2 trial,” Lancet, 2025 (advance online publication).
- S. J. Nicholls, C. M. Ballantyne, and colleagues (CORALreef Lipids Investigators), “Efficacy and safety of enlicitide, an oral PCSK9 inhibitor, in adults with hypercholesterolemia: the Phase 3 CORALreef Lipids trial,” N. Engl. J. Med., 2025 (publication pending).
- M. Bhandari, S. M. Nissen, and colleagues, “How low can you go? New evidence supports no lower bound to low-density lipoprotein cholesterol level in secondary prevention,” Circulation, vol. 147, no. 19, pp. 1433–1435, May 2023, doi: 10.1161/CIRCULATIONAHA.123.064041.
- P. M. Ridker, N. Mora, L. Rose, and JUPITER Trial Study Group, “Percent reduction in LDL cholesterol following high-intensity statin therapy: potential implications for guidelines and for the prescription of emerging lipid-lowering agents,” Eur. Heart J., vol. 37, no. 17, pp. 1373–1379, May 2016, doi: 10.1093/eurheartj/ehw046.
- P. Libby, P. M. Ridker, and G. K. Hansson, “Progress and challenges in translating the biology of atherosclerosis,” Nature, vol. 473, no. 7347, pp. 317–325, May 2011, doi: 10.1038/nature10146.
- S. Tsimikas, “A test in context: lipoprotein(a): diagnosis, prognosis, controversies, and emerging therapies,” J. Am. Coll. Cardiol., vol. 69, no. 6, pp. 692–711, Feb. 2017, doi: 10.1016/j.jacc.2016.11.042.
- S. Tsimikas, H.-K. Gordts, C. Nora, C. Yeang, and J. L. Witztum, “Statin therapy increases lipoprotein(a) levels,” Eur. Heart J., vol. 41, no. 24, pp. 2275–2284, Jun. 2020, doi: 10.1093/eurheartj/ehz310.
- P. M. Ridker, B. M. Everett, T. Thuren, J. G. MacFadyen, W. H. Chang, C. Ballantyne, F. Fonseca, J. Nicolau, W. Koenig, S. D. Anker, J. J. P. Kastelein, J. H. Cornel, P. Pais, D. Pella, J. Genest, R. Cifkova, A. Lorenzatti, T. Forster, Z. Kobalava, L. Vida-Simiti, M. Flather, H. Shimokawa, H. Ogawa, M. Dellborg, P. R. F. Rossi, R. P. T. Troquay, P. Libby, R. J. Glynn, and CANTOS Trial Group, “Antiinflammatory therapy with canakinumab for atherosclerotic disease,” N. Engl. J. Med., vol. 377, no. 12, pp. 1119–1131, Sep. 2017, doi: 10.1056/NEJMoa1707914.
- A. Daugherty, S. E. Tall, M. J. Daemen, E. Falk, E. M. Fisher, E. Garcia-Cardena, A. R. Lusis, A. P. Owens III, M. E. Rosenfeld, and I. Tabas, “Recommendation on design, execution, and reporting of animal atherosclerosis studies: a scientific statement from the American Heart Association,” Arterioscler. Thromb. Vasc. Biol., vol. 37, no. 9, pp. e131–e157, Sep. 2017, doi: 10.1161/ATV.0000000000000062.
- M. J. Budoff, S. Achenbach, R. S. Blumenthal, J. J. Carr, J. G. Goldin, P. Greenland, A. D. Guerci, J. A. Lima, D. J. Rader, G. D. Rubin, L. J. Shaw, S. E. Wiegers, American Heart Association Committee on Cardiovascular Imaging and Intervention, American Heart Association Council on Cardiovascular Radiology and Intervention, American Heart Association Committee on Cardiac Imaging, “Assessment of coronary artery disease by cardiac computed tomography: a scientific statement from the AHA,” Circulation, vol. 114, no. 16, pp. 1761–1791, Oct. 2006, doi: 10.1161/CIRCULATIONAHA.106.178458.
- H. S. Hecht, “Coronary artery calcium scanning: past, present, and future,” JACC Cardiovasc. Imaging, vol. 8, no. 5, pp. 579–596, May 2015, doi: 10.1016/j.jcmg.2015.02.006.
- K. Nasir, M. B. Rivera, R. Blankstein, M. J. Blaha, A. D. Choi, R. S. Blumenthal, M. G. Silverman, A. Dardari, J. Berman, J. Yeboah, M. J. Budoff, J. J. Carr, and M. Cainzos-Achirica, “Implications of coronary artery calcium testing for primary prevention,” J. Am. Coll. Cardiol., vol. 66, no. 15, pp. 1657–1668, Oct. 2015, doi: 10.1016/j.jacc.2015.07.066.
- S. E. Nissen, S. J. Nicholls, I. Sipahi, P. Libby, J. S. Raichlen, C. M. Ballantyne, J. Davignon, R. Erbel, J. C. Fruchart, J.-C. Tardif, P. Schoenhagen, T. Crowe, V. Cain, K. Wolski, M. Goormastic, and E. M. Tuzcu, “Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial,” JAMA, vol. 295, no. 13, pp. 1556–1565, Apr. 2006, doi: 10.1001/jama.295.13.jpc60002.
- S. E. Nissen, S. J. Nicholls, K. Wolski, R. Nesto, S. Kupfer, A. Perez, H. Jure, R. De Larochellière, C. S. Staniloae, K. Mavromatis, J. Saw, B. Hu, A. M. Lincoff, E. M. Tuzcu, and PERISCOPE Investigators, “Effect of rosuvastatin versus atorvastatin on progression of coronary atherosclerosis in patients with coronary artery disease (SATURN),” N. Engl. J. Med., vol. 365, no. 22, pp. 2078–2087, Dec. 2011, doi: 10.1056/NEJMoa1110874.
- S. J. Nicholls, R. Puri, T. Anderson, C. M. Ballantyne, L. Cho, J. J. P. Kastelein, W. Koenig, R. Somaratne, H. Kassahun, J. Yang, S. M. Wasserman, R. Scott, I. Ungi, J. Podolec, A. O. Ophuis, J. H. Cornel, M. Borgman, D. M. Brennan, and S. E. Nissen, “Effect of evolocumab on progression of coronary disease in statin-treated patients: the GLAGOV randomized clinical trial,” JAMA, vol. 316, no. 22, pp. 2373–2384, Dec. 2016, doi: 10.1001/jama.2016.16951.
- A. V. Khera, E. S. Everett, M. G. Caulfield, F. E. Hantgan, J. Deasy, R. K. Scott, P. S. Sever, J. Barton, L. Heller, A. Nambi, and M. S. Sabatine, “Evolocumab, plaque regression, and clinical outcomes,” J. Am. Coll. Cardiol., vol. 75, no. 9, pp. 1037–1050, Mar. 2020, doi: 10.1016/j.jacc.2019.12.052.
- D. J. Rader and G. K. Hovingh, “HDL and cardiovascular disease,” Lancet, vol. 384, no. 9943, pp. 618–625, Aug. 2014, doi: 10.1016/S0140-6736(14)61217-4.
- R. C. Thompson, A. H. Allam, G. P. Lombardi, L. S. Wann, M. L. Sutherland, J. D. Sutherland, M. A. Soliman, B. Frohlich, D. T. Mininberg, J. M. Monge, C. M. Vallodolid, S. L. Cox, G. A. El-Maksoud, I. Badr, M. I. Miyamoto, A. el-Halim Nur el-Din, J. Narula, C. E. Finch, and G. S. Thomas, “Atherosclerosis across 4000 years of human history: the Horus study of four ancient populations,” Lancet, vol. 381, no. 9873, pp. 1211–1222, Apr. 2013, doi: 10.1016/S0140-6736(13)60598-X.
- H. Kaplan, R. C. Thompson, B. C. Trumble, L. S. Wann, A. H. Allam, B. Beheim, B. Frohlich, M. L. Sutherland, J. D. Sutherland, J. Stieglitz, D. E. Rodriguez, D. E. Michalik, C. J. Rowan, G. P. Lombardi, R. Bedi, C. E. Garcia, J. K. Min, J. Narula, C. E. Finch, M. Gurven, and G. S. Thomas, “Coronary atherosclerosis in indigenous South American Tsimane: a cross-sectional cohort study,” Lancet, vol. 389, no. 10080, pp. 1730–1739, Apr. 2017, doi: 10.1016/S0140-6736(17)30752-3.