Your Heart Disease Started 20 Years Ago: Here’s How to Stop It
-
Introduction: The Quiet Neighbor with a Big Secret
Most people think a heart attack is like a lightning strike. One minute you are fine, and the next, everything changes. But science shows that heart disease is not a sudden accident. It is more like a slow story that takes forty or fifty years to write.
Doctors call the early part of this story “subclinical atherosclerosis.” That is a very big name for a simple problem: “gunk” building up inside the pipes of your body. Imagine the plumbing in your house. Over many years, hair, soap, and grease slowly stick to the inside of the pipes. You do not notice it today. You do not notice it tomorrow. But the gunk is growing. Usually, we only call the plumber when the sink stops draining or a pipe bursts. In our bodies, we often wait until a heart attack happens to worry about our “plumbing.” By then, the gunk has been there for decades.
You can also think of your arteries like a brand-new non-stick frying pan. When the pan is new, nothing sticks to it. But as the years go by, high heat and metal spoons scratch the surface. Once that “non-stick” coating is gone, food starts to burn and stick to the pan. Your arteries are the same. They start out smooth and slippery. But life causes “scratches” on the inside. This lets the gunk—mostly a mix of fat and cholesterol—start to pile up. The good news is that if we understand how this clock works, we can actually learn how to turn it back.
-
It Starts Before You Were Born (The Most Surprising Fact)
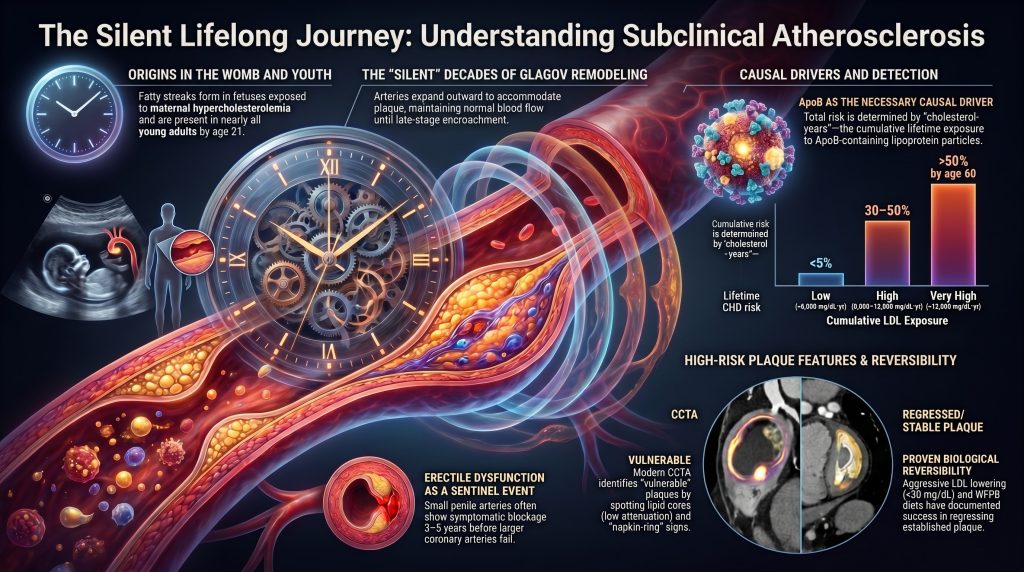
We used to think that heart disease was only for old people. We thought it was something you “got” when you retired. But famous studies have changed how we think. Doctors looked at the arteries of children and even babies who died in accidents. They found something shocking. The very first signs of gunk, which they call “fatty streaks,” can start before a baby is even born.
If a mother has very high cholesterol while she is pregnant, it can actually “program” the baby’s pipes to collect gunk more easily later in life. By the time children are only two years old, many already have these streaks starting in their main pipes.
“The earliest documented atherosclerotic lesion in humans is fetal… fatty streaks form in the aortic intima of fetuses, with intimal accumulation of LDL and its oxidation preceding monocyte recruitment.”
By the time most kids are in high school, these streaks are present in almost everyone. This means that heart disease is a life-long journey. It is a silent clock that starts ticking long before we ever think about our health. We are not “fine” until we are 50. Instead, we are either building up gunk or keeping our pipes clean every single day, starting from day one.
-
The “Sunburn” Effect: Why Total Exposure is the Only Number That Matters
Imagine you go to the beach. If you stay in the hot sun for ten minutes, you probably won’t get a sunburn. But if you stay out for six hours in mild sun, you will definitely burn. It is the total amount of sun over time that causes the damage. Your heart works exactly the same way.
Scientists use a special measurement called “Cholesterol-Years.” Think of it like “pack-years” for someone who smokes. If you smoke one pack a day for 40 years, your lungs are in much worse shape than someone who smoked five packs a day for just one week. For your arteries, having slightly high cholesterol for 40 years is much more dangerous than having very high levels for just a few years.
To understand this, we need to talk about “ApoB.” Think of ApoB as the number of “delivery trucks” on the road. These trucks carry “cargo,” which is the cholesterol. Most doctors only weigh the cargo (this is called LDL-C). But the real danger comes from the trucks themselves. Each truck has a chance to crash into your artery wall and get stuck. If you have too many trucks on the road for too many years, your “bucket” eventually overflows.
There is a specific “tipping point” that scientists have found. If your “total exposure” reaches a score of 5,000, that is when the “leaks” and “clogs” usually start to happen. For example, if your LDL level is 125 and you stay at that level for 40 years, you hit that 5,000 mark (125 times 40). By then, the damage is often done. This is why it is so important to keep your “truck count” low as early as possible. If the bucket never fills up, it can never overflow.
-
The Geography of the Body: Why the “Curves” Get Clogged First
If you look at a river, the water moves fast and clear in the straight parts. But look at the bends and curves. That is where the water slows down and swirls around. Because the water is slower there, mud and silt settle at the bottom.
Your blood vessels work just like that river. In the straight parts of your pipes, blood flows smoothly. This smooth flow actually helps keep the “non-stick coating” of your arteries healthy. But your body’s plumbing has many sharp turns and branches.
The most famous spot for this is an artery called the LAD (the Left Anterior Descending artery). This is a main pipe that feeds the front of your heart. It has a very sharp “take-off” angle, like a sudden exit on a highway. Because the turn is so sharp, the blood becomes “disturbed.” It swirls and gets messy. These “curves” are the zones where the gunk has the easiest time sticking to the walls. It isn’t random where the clogs happen; it is a matter of where the “mud” settles in the river of your blood.
-
The Canary in the Coal Mine: Early Warning Signs You Can’t Ignore
Even though this disease is called “silent,” it actually leaves clues if you know where to look. One of the biggest clues is how your body “hides” the gunk at first. This is called the Glagov Phenomenon.
Imagine you are wearing a belt that is getting too tight because you are gaining weight. To hide it, you might just loosen the belt by one or two notches. Your waist gets bigger, but your pants still fit the same. Your arteries do the same thing! When gunk starts to build up, the artery actually expands outward to keep the tunnel inside the same size. This is why a person can feel totally “fine” and pass a stress test even when they have lots of gunk. The artery is hiding the mess by stretching outward. But eventually, the “belt” can’t be loosened anymore. That is when the pipe finally starts to narrow and the trouble begins.
Before the big pipes in your heart clog, the tiny pipes elsewhere give out. This is the “Artery-Size Hypothesis.” The pipes in your heart are about 3 to 4 millimeters wide. But the pipes that control blood flow for male erections are only 1 to 2 millimeters wide. Because they are smaller, they clog much sooner.
“Erectile dysfunction (ED) is often the first clinical manifestation of systemic vascular disease… the cavernosal circulation reaches the threshold for symptomatic compromise years before the coronary circulation does.”
Because of this, if a man has trouble with ED, it is often a 3-to-5-year warning sign that a heart attack might be coming. It is the “canary in the coal mine.” Other signs include “slower thinking” or getting tired much faster during a workout. If the tiny pipes in your brain or muscles are getting gunked up, you won’t have the “reserve” energy you used to have.
-
Hidden Hazards: The Risks Your Standard Test Might Miss
Sometimes, people eat well and have “normal” test results, but they still have heart issues. This happens because standard tests miss two “hidden hazards.”
The first is a “genetic hitchhiker” called Lipoprotein(a), or Lp(a). About one in five people (20% of the world) are born with this. It is like an extra-sticky delivery truck that carries “extra-toxic” chemicals. These chemicals irritate your pipes and make them “angry.” Regular cholesterol tests do not see this truck at all. You only need to test for this once in your life to know if you have it. If you do, you have to be much more careful with your health.
The second hazard is “inflammation.” Think of heart health like a fire. To have a fire, you need “fuel” (the gunk) and a “spark” (inflammation). Inflammation is when your body is on “high alert” or “angry.” We can measure this with a test called hs-CRP. Think of hs-CRP as a thermometer that tells us how “hot” the fire in your arteries is burning. If the thermometer is high, the gunk in your pipes is much more likely to cause a “burst.”
-
Good News: You Can Turn Back the Clock (The Power of Reversal)
For a long time, doctors thought that once the gunk was in your pipes, it was there forever. But we now know that is wrong! If you get your “truck count” (ApoB) very low and put out the “fire” of inflammation, your body can actually start to heal.
“Subclinical atherosclerosis is, when addressed early and aggressively, a reversible disease. Plaque can be stabilized, fibrous caps thickened, and event rates reduced.”
There are amazing “Success Stories” in the science:
- The Lifestyle Heart Trial: Dean Ornish showed that when people ate a whole-food, plant-based diet and managed their stress, their clogs actually got smaller. Their bodies started “cleaning” the pipes.
- The “Tough Scab” Discovery: New studies like HUYGENS and PACMAN-AMI show that intensive treatment can change the type of gunk you have. Instead of being soft and “fragile”—like a pimple that is ready to pop—the gunk develops a “thick cap.” This cap is like a thick, tough scab over a scrape. It makes the gunk stable and safe so it doesn’t cause a heart attack.
- Intensive Cleaning: When people get their cholesterol very low, the body can actually pull the gunk out of the artery walls and “shrink” the buildup.
This means you are not stuck with the plumbing you have today. Your choices—what you eat, how you move, and the modern medicines you use—can literally turn back the clock.
-
Conclusion: Your Future Self is Watching
Your heart health is like a lifelong savings account. Every day that you keep your pipes clean, you are making a deposit into your future. We now know that the “silent clock” starts early, even before we are born. But we also know we have the tools to slow it down or even wind it back.
Heart health is about more than just avoiding a “big event” later. It is about keeping your brain sharp, your energy high, and your body working correctly today. If you wait until the “pipes leak” to care about your heart, you have missed forty years of opportunity.
If you could see inside your pipes today, would you change how you treat them tomorrow? The good news is that you don’t have to wait for a disaster to start. By understanding the “gunk,” the “trucks,” and the “river bends,” you can take control of your story. Your future self is counting on the choices you make right now.
DEEP DIVE
Pathophysiological Progression and Systemic Functional Impact of Subclinical Atherosclerosis
A Life-Course Analysis from Youth to Mid-Life
Abstract
Atherosclerotic cardiovascular disease (ASCVD) remains the leading cause of mortality worldwide, and its clinical manifestations represent the late-stage culmination of a biological trajectory that begins decades earlier — often in fetal life and certainly by the second decade. This review integrates structural vascular biology, hemodynamics, lipid causality, inflammation, sex-specific phenotypes, neurodegeneration, and lifestyle reversibility into a unified framework for understanding subclinical atherosclerosis as an active, progressive, and modifiable disease. We examine regional heterogeneity in arterial wall architecture and the role of vasa vasorum, the divergence of large-artery atherosclerosis from cerebral small vessel disease, the hemodynamic determinants of plaque localization, and the longitudinal evidence from PDAY, Bogalusa, CARDIA, MESA, and the FELIC fetal series that anchor disease initiation in early life. We synthesize Mendelian-randomization, population-genetic, and intervention-trial evidence establishing apolipoprotein B–containing lipoproteins as the necessary causal driver of atherogenesis, and we situate Lp(a), inflammation, clonal hematopoiesis, endothelial dysfunction, microvascular rarefaction, and arterial stiffening within this framework. We close with the imaging and biomarker tools that allow detection during the long pre-clinical window, the sex-specific phenotypes that have historically been under-recognized, and the trial evidence — most prominently the Lifestyle Heart Trial, the Esselstyn case series, STARS, and the IVUS-documented statin and PCSK9-inhibitor regression trials (REVERSAL, ASTEROID, SATURN, GLAGOV, PACMAN-AMI, HUYGENS) — supporting the conclusion that subclinical atherosclerosis is, when addressed early and aggressively, a reversible disease.
Keywords
Subclinical atherosclerosis; apolipoprotein B; lipoprotein(a); endothelial dysfunction; vasa vasorum; cerebral small vessel disease; coronary artery calcium; pulse wave velocity; plaque regression; whole-food plant-based diet.
1. Introduction
The emergence of clinical ASCVD is the culmination of a silent, multi-decadal biological trajectory that often begins in the second decade of life — and in the case of fetuses exposed to maternal hypercholesterolemia, even before birth. Subclinical atherosclerosis refers to the presence of structural arterial wall alterations — including intimal thickening, lipoprotein retention, foam-cell accumulation, and early plaque formation — that precede overt symptomatic manifestations such as angina, myocardial infarction, ischemic stroke, peripheral claudication, or vascular dementia. This silent progression is governed by a tightly coupled interplay between hemodynamic forces, structural variations in the arterial wall, lipoprotein flux, immune activation, and metabolic stress.
Although traditionally framed as a disease of senescence, longitudinal cohort studies and autopsy data have fundamentally shifted the focus toward young adults and adolescents, revealing that subtle everyday functional deficits — ranging from diminished cognitive processing speed to impaired renal and erectile reserve — emerge long before the traditional clinical thresholds for diagnosis are met. The most rigorous formulation of this life-course concept is the cumulative-exposure or “cholesterol-years” model, in which the integral of plasma apoB-particle concentration over time predicts both the timing and severity of clinical events [7], [9].
This review provides a comprehensive synthesis of the pathophysiology, anatomical distribution, and functional consequences of subclinical vascular disease across the human arterial tree, with particular attention to (i) the divergent biology of large-artery atherosclerosis and cerebral small vessel disease; (ii) the causal centrality of apoB-containing lipoproteins and the contribution of Lp(a), inflammation, and clonal hematopoiesis; (iii) sex-specific phenotypes; (iv) emerging detection modalities; and (v) the trial-based evidence that subclinical disease is reversible when addressed early and aggressively.
2. Apolipoprotein B–Containing Lipoproteins as the Necessary Causal Driver
2.1 The Mendelian-Randomization Synthesis
Mendelian randomization (MR) provides one of the most rigorous forms of causal inference available outside randomized controlled trials. By exploiting the random allocation of alleles at conception, MR studies estimate the lifelong effect of a genetically determined exposure unconfounded by reverse causation, lifestyle covariates, or measurement error. Ference and colleagues analyzed 50 polymorphisms across nine LDL-related genes in 312,321 participants and established that each 1 mmol/L (≈38.7 mg/dL) genetically lower LDL-C confers a relative risk reduction of approximately 54.5 percent for coronary heart disease — an effect roughly three times larger per unit LDL than that of statins initiated in mid-life [1].
The dose-response relationship is log-linear with no detectable threshold, supporting a cumulative-exposure model. A second MR study using polymorphisms in PCSK9, HMGCR, and NPC1L1 demonstrated that risk reduction tracks closely with the magnitude and duration of LDL-C lowering, supporting the conclusion that integrated lifetime exposure to apoB-containing lipoproteins is a central determinant of atherogenesis, while the specific molecular pathway through which LDL-C is lowered is less important for ASCVD risk reduction [3]. A 2017 European Atherosclerosis Society Consensus Statement reviewed more than 200 genetic, prospective epidemiologic, and randomized intervention studies and concluded that LDL fulfills the Bradford Hill criteria for causality in ASCVD [4], and the 2020 EAS update extended this conclusion to apoB-containing lipoproteins generally [19].
2.2 ApoB as the Unifying Particle
Each major atherogenic lipoprotein particle — VLDL, IDL, LDL, Lp(a), and chylomicron remnants — carries a single apolipoprotein B molecule: apoB-100 for hepatically derived particles and apoB-48 for intestinal remnants. ApoB therefore counts atherogenic particles, whereas LDL-C measures a cargo whose ratio to the carrier varies with metabolic state. In the UK Biobank analysis of 389,529 participants, apoB was the dominant predictor of myocardial infarction; LDL-C and non-HDL-C lost statistical significance after adjustment for apoB [5]. In states of discordance — where LDL-C is normal but particle number is high, as in metabolic syndrome and insulin resistance — apoB unmasks substantial atherogenic risk that an LDL-C-only strategy would miss [6].
2.3 Cumulative Exposure and the “Cholesterol-Years” Concept
The cumulative-exposure model treats ASCVD risk as a function of the integral of plasma LDL-C concentration over time, analogous to “pack-years” for tobacco. Several analyses support a dose–time relationship between LDL-C exposure and ASCVD events. The “threshold” for clinically apparent disease in men has been described in some analyses as approximately 5,000 mg/dL·years — for example, LDL 125 mg/dL × 40 years, corresponding to age ~50 in modern Western populations — but the exact LDL-year value at which clinical disease emerges is model-dependent and should be presented as illustrative rather than as a universal biological cutoff [7], [9].
The “LDL plaque-years” framework reframes prevention quantitatively around cumulative burden. For example, lifelong LDL of 70 mg/dL × 80 years yields ≈5,600 mg/dL·years; LDL 130 mg/dL × 50 years yields ≈6,500 mg/dL·years; and untreated heterozygous familial hypercholesterolemia patients with LDL of 250 mg/dL exceed common illustrative thresholds by their early thirties. These calculations are useful for communicating dose–time biology and for explaining why untreated heterozygous FH carries a high lifetime CHD risk, but they should not be presented as validated individual-risk thresholds [7], [8], [9].
| Category | Cumulative LDL exposure | Approximate trajectory | Illustrative lifetime CHD risk |
| Low | <5,000 mg/dL·yr | LDL 70 × 70 yr | <5% |
| Intermediate | 5,000–8,000 mg/dL·yr | LDL 100 × 60 yr | 10–20% |
| High | 8,000–12,000 mg/dL·yr | LDL 130 × 65 yr | 30–50% |
| Very high | >12,000 mg/dL·yr | LDL 190 × 65 yr (HeFH) | >50% by age 60 |
Table 1. Illustrative cumulative LDL-cholesterol exposure categories and approximate lifetime coronary heart disease risk. Adapted from Ference et al. and Domanski et al. [7], [8], [9]. These categories are illustrative communication aids, not guideline-validated individual-risk thresholds.
2.4 Tsimane and Hadza: The Natural Experiment of Lifelong Low LDL
Kaplan and colleagues examined CAC scores in 705 Tsimane forager-horticulturalists of the Bolivian Amazon aged 40 to 94. The mean LDL-C was 91 mg/dL, and 85 percent of adults aged 40 and older had a CAC of zero, compared with approximately 50 percent of US adults of similar age in MESA. Sixty-five percent of those over 75 had a CAC of zero — the lowest level of coronary atherosclerosis ever measured in any population — and only 8 percent had a CAC ≥100, versus more than 50 percent in age-matched US populations [10].
This natural experiment supports the conclusion that maintaining relatively low LDL-C across the life course, combined with high physical activity and minimal smoking, is associated with very low prevalence of coronary calcification. It should not be framed as proving that LDL-C ≤90 mg/dL virtually eliminates coronary atherosclerosis, because the Tsimane phenotype reflects multiple lifelong exposures and not LDL-C in isolation. The Hadza of Tanzania have been described as a physically active hunter-gatherer population with favorable cardiometabolic features in available studies, but direct CAC imaging has not been performed in this group; conclusions about the absence of clinical coronary disease in the Hadza are therefore inferential rather than direct [11].
2.5 Genetic Lifelong Low LDL: PCSK9 and ANGPTL3
Cohen and colleagues identified naturally occurring loss-of-function mutations in PCSK9 (R46L, Y142X, C679X) that lower LDL by 15–40 percent across the life course. Carriers of selected variants exhibited large reductions in CHD events, with the most striking estimates — including reductions approaching 88 percent for some variants — reflecting effect sizes that vary by variant and ancestry [12]. Such effect sizes far exceed what any pharmacologic intervention initiated in mid-life can achieve. Similarly, homozygous loss-of-function variants in ANGPTL3 produce hypobetalipoproteinemia with very low LDL and triglycerides; epidemiologic series describe markedly reduced ASCVD risk and favorable lipid profiles in such carriers, although event rates have not been quantified in randomized cohorts [13].
These genetic experiments establish a useful boundary condition: when lifelong apoB-particle burden is genetically low, atherosclerosis appears markedly suppressed even without pharmacologic intervention. Pharmacologic mimicry of these phenotypes — through PCSK9 monoclonal antibodies, siRNA agents, and ANGPTL3 inhibitors — has demonstrated additive event reduction on top of statin therapy. Analyses of FOURIER and the FOURIER open-label extension support progressively lower event rates among patients achieving very low LDL, including LDL <20 mg/dL, without an excess safety signal over available follow-up [18].
2.6 Why Sustained Very Low LDL Markedly Reduces Atherogenesis
Endothelial transcytosis of LDL into the intima is concentration-dependent, and proteoglycan-mediated retention — the response-to-retention paradigm of Tabas, Williams, and Borén — becomes less likely as apoB-particle flux falls [83]. Newborns have LDL-C ≈30 mg/dL, and atherosclerotic lesions are not typically detected at this stage; this should be interpreted as evidence that very low lifelong LDL-C is incompatible with the early steps of atherogenesis on a population scale, rather than as a claim that atherosclerosis is biologically impossible at any single LDL-C value. Cohorts with lifelong LDL <70 mg/dL (PCSK9 LOF, ANGPTL3 LOF, treated FH) demonstrate marked reductions in event rates, with IVUS-documented plaque regression at on-treatment LDL <60 mg/dL across the REVERSAL, ASTEROID, SATURN, and GLAGOV trials [14], [15], [16], [17].
FOURIER-OLE extended evolocumab follow-up for a median of approximately 5 years and reported continued event reduction at on-treatment LDL <20 mg/dL with no excess safety signal over available follow-up [18]. A formal demonstration of dose-response without plateau at these very low LDL levels has not been firmly established, but available data are consistent with continued benefit and no offsetting toxicity through the lowest LDL achieved in trial populations to date.
2.7 Acknowledging and Rebutting the LDL-Skeptic Position
A small group of authors — most prominently Ravnskov, Diamond, and Kendrick — have argued that LDL is non-causal, citing observational studies in elderly populations in which LDL appears non-predictive (the so-called “lipid paradox”). The standard rebuttals are well established. First, reverse causality dominates in late life: chronic illness, malabsorption, and frailty lower LDL, biasing the LDL–mortality association in cross-sectional analyses. Second, survivor bias selects for genetically protected individuals among those reaching age 80 with high LDL. Third, attenuation of relative risk with age does not imply attenuation of absolute risk; the absolute event rate increases dramatically with age. Fourth, the genetic and pharmacologic evidence is mutually corroborative across approximately twenty independent lines of investigation, satisfying triangulation criteria for causality [4], [6], [19]. The skeptic position rests almost entirely on observational data while ignoring the convergent genetic and randomized-trial evidence.
3. Structural Determinants and the Role of Vasa Vasorum in Atherogenesis
3.1 Critical Depth and the Lamellar Unit
In large-caliber systemic arteries, the metabolic demands of the thick wall exceed the capacity for simple oxygen diffusion from the lumen. The “critical depth” is defined as the physiological limit of oxygen and nutrient diffusion from luminal blood, established by Geiringer at approximately 0.5 mm (≈500 μm), or roughly 29–30 lamellar units of the medial wall [78], [79]. Each lamellar unit consists of an elastic lamina and its associated layer of smooth-muscle cells and extracellular matrix, approximately 15 μm thick. Wall segments thicker than this threshold require an intrinsic microvascular network — the vasa vasorum — to maintain viability of the outer media and adventitia.
Vasa vasorum are categorized into vasa vasorum interna, which arise directly from the arterial lumen, and vasa vasorum externa, which originate from remote branches and penetrate the adventitia. Geiringer’s investigations established that vasa vasorum are abundant in the adventitia and outer third of the media, while the inner ≈0.5 mm (≈30 lamellar units) of the wall remains avascular and is supplied solely by luminal diffusion [79]. When the wall thickens because of atherosclerotic plaque or hypertensive hyperplasia, the diffusion distance increases, generating a hypoxic environment in the deeper layers. Hypoxia triggers HIF-1α–dependent angiogenic signaling, leading to proliferation of vasa vasorum that can penetrate the internal elastic lamina and enter the plaque itself, where they serve both as conduits for inflammatory cells and as fragile sources of intraplaque hemorrhage.
3.2 Extracranial vs. Intracranial Vascular Nourishment
The intracranial vasculature presents a unique architectural profile compared with the systemic circulation. Healthy intracranial arteries are characterized by a thinner tunica media, less abundant adventitia, and a relative paucity of elastic fibers; many lack a well-defined external elastic lamina. A primary distinguishing feature is that intracranial vessels are bathed in nutrient-rich cerebrospinal fluid, which provides metabolic support through external diffusion and partly compensates for the relative absence of vasa vasorum early in life.
Modern autopsy and imaging studies have refined the historical view that vasa vasorum are absent in the brain. In one autopsy series of 50 cases, vasa vasorum were identified in 72 percent of patients, localized primarily in the tunica adventitia. Their distribution is markedly non-uniform: vasa vasorum are more frequently found in proximal segments — the vertebral arteries, basilar artery, and intracranial portion of the internal carotid artery — than in distal segments such as the middle cerebral or anterior cerebral arteries. Although atherosclerosis can occur in the absence of vasa vasorum, their development is strongly associated with the progression of intracranial disease: in the intracranial vertebral artery, the presence of adventitial vasa vasorum correlates with greater plaque load, denser intraplaque calcification, and more severe luminal stenosis.
| Vessel type | Wall thickness | Vasa vasorum (early life) | Nutritional source |
| Aorta / large systemic | >1.0 mm (often 1.5–2.0 mm) | Abundant in adventitia and outer media | Luminal diffusion + VV |
| Extracranial carotid | ≈0.6–1.0 mm | Present in adventitia | Luminal diffusion + VV |
| Intracranial ICA / VA | ≈0.2–0.3 mm | Sparse; mostly proximal segments | Luminal diffusion + CSF |
| Distal MCA / ACA | <0.2 mm; lacks EEL | Absent or rare | Luminal diffusion + CSF |
| Penetrating arterioles | 40–200 μm diameter | Absent | Luminal diffusion + interstitial fluid |
Table 2. Comparative architecture of arterial wall thickness, vasa vasorum density, and nutritional source across the systemic and cerebral circulations. EEL, external elastic lamina; ICA, internal carotid artery; VA, vertebral artery; MCA, middle cerebral artery; ACA, anterior cerebral artery; VV, vasa vasorum; CSF, cerebrospinal fluid.
4. Hemodynamic Forces and Plaque Localization
4.1 Laminar versus Disturbed Shear Stress
The focal nature of atherosclerosis is dictated by the interaction between blood flow and arterial geometry. The endothelium serves as a mechanosensor, translating physical stress into biological signaling through primary cilia, integrins, glycocalyx-mediated mechanotransduction, and ion channels (notably Piezo1 and TRPV4). In straight arterial segments, flow is laminar, generating high, unidirectional wall shear stress (typically 1–7 Pa, or 10–70 dyn/cm²). This environment maintains an atheroresistant endothelial phenotype characterized by sustained activation of endothelial nitric oxide synthase (eNOS), phosphorylation of KLF2 and KLF4 transcription factors, and suppression of NF-κB signaling, with the result that nitric oxide production is high, leukocyte adhesion is suppressed, and intimal permeability remains low [85].
In contrast, at branch points, bifurcations, and areas of high curvature, flow becomes “disturbed.” These atheroprone zones experience low time-averaged wall shear stress (often <0.4 Pa) and high oscillatory shear index, with the direction of frictional force reversing during the cardiac cycle. In disturbed-flow regions, the endothelium undergoes a phenotypic switch: tight junctions loosen, allowing increased transcytosis of LDL into the intima; expression of VCAM-1, ICAM-1, E-selectin, and MCP-1 captures circulating monocytes and T-cells; reactive oxygen species generation rises through NADPH oxidase activation; and the protective KLF2/eNOS axis is suppressed.
The seminal computational fluid dynamic study by Ku and colleagues at the human carotid bifurcation demonstrated tight spatial concordance between low-shear regions and intimal thickening — the founding empirical study of the hemodynamic theory of atherogenesis [71]. This pattern recurs at every branching point of the arterial tree: the proximal segments of the LAD and LCx, the carotid bulb, the abdominal aortic bifurcation, and the renal artery ostia all exhibit this geometry-dependent vulnerability.
4.2 Cellular Behavior in the Plaque Microenvironment
Once retained in the intima, LDL undergoes oxidative modification by myeloperoxidase, lipoxygenase, and reactive oxygen species. Oxidized LDL is recognized by scavenger receptors (CD36, SR-A) on resident and newly recruited macrophages, which internalize it and become foam cells. In early subclinical lesions, foam-cell death is balanced by efferocytosis — the clearance of apoptotic cells by neighboring macrophages — but as the microenvironment becomes increasingly toxic, efferocytosis fails, apoptotic and necrotic debris accumulates, and a necrotic core forms. Vascular smooth-muscle cells (VSMCs) simultaneously switch from a contractile to a synthetic phenotype, migrating from the media into the intima where they secrete a collagen-rich fibrous cap that initially stabilizes the lesion. The balance between cap-thickening repair and core-expanding inflammation defines whether a plaque remains stable or progresses to vulnerability [82], [83], [84].
5. Endothelial Dysfunction as the Earliest Detectable Lesion
Endothelial dysfunction precedes any structural lesion detectable by carotid intima-media thickness (CIMT), coronary artery calcium scoring, or angiography. It is functional, dynamic, and partially reversible — and therefore represents the earliest practical window for primordial intervention.
5.1 Flow-Mediated Dilation
Brachial flow-mediated dilation (FMD), measured by ultrasound after a 5-minute forearm cuff occlusion, quantifies endothelium-dependent (largely nitric-oxide-mediated) vasodilation. Lower FMD is associated with increased cardiovascular risk, but FMD is protocol-dependent and no single universal cutoff applies across laboratories; values below approximately 5–7 percent are commonly treated as abnormal in research contexts. In the Multi-Ethnic Study of Atherosclerosis, FMD added independent prognostic information beyond Framingham risk and CIMT [33]. FMD is impaired in subjects with even mildly elevated LDL, insulin resistance, untreated hypertension, or chronic exposure to particulate air pollution.
5.2 Reactive Hyperemia Index
The reactive hyperemia index (RHI), measured non-invasively by digital plethysmography (EndoPAT), reflects microvascular endothelial function in the fingertip after reactive hyperemia. An RHI threshold around 1.67 has been used to identify coronary endothelial dysfunction or early coronary atherosclerosis in selected cohorts; sensitivity and specificity depend on the population and the reference standard [34]. RHI is operator-independent, has good reproducibility, and has been used as an outcome in lifestyle and pharmacologic intervention trials.
5.3 Glycocalyx Degradation
The endothelial glycocalyx is a 0.5–3 μm gel-like surface layer composed of proteoglycans (heparan sulfate, chondroitin sulfate), glycoproteins (syndecan-1), and adsorbed plasma proteins. It modulates LDL transcytosis, leukocyte rolling, and shear-stress mechanotransduction. Glycocalyx degradation — driven by oxidized LDL, hyperglycemia, TNF-α, and reactive oxygen species — exposes adhesion molecules and increases intimal lipoprotein flux. Glycocalyx thinning, detectable by sublingual sidestream dark-field imaging and by elevated plasma syndecan-1 and hyaluronan, occurs before measurable FMD impairment and is among the earliest detectable abnormalities in subclinical disease [35], [36].
5.4 Microvascular Dysfunction Preceding Macrovascular Disease
Coronary flow reserve (CFR) below 2.0 by positron emission tomography, below 2.5 by transthoracic Doppler, or below 2.0 by cardiac magnetic resonance is independently predictive of cardiovascular events even in the absence of obstructive epicardial disease. Microvascular endothelial dysfunction can occur with normal coronary angiograms, providing the substrate for the syndrome of ischemia with non-obstructive coronary arteries (INOCA), discussed in Section 8 [37].
6. Anatomical Mapping of Subclinical Vascular Disease
Atherosclerosis is a systemic but non-uniform disease, favoring specific anatomical sites characterized by complex geometry and disturbed flow. Precise mapping across vascular beds reveals the predictable, geometry-dependent pattern of plaque localization.
6.1 Cerebrovascular and Cervical Beds
In the neck, the carotid bifurcation and the proximal internal carotid artery are the primary sites for early plaque development, owing to flow separation, recirculation, and low oscillatory shear at the carotid bulb. Within the cranium, disease is most frequently observed in the intracranial ICA (carotid siphon) and the proximal segments of the major branches of the circle of Willis. Autopsy series indicate that intracranial atherosclerosis lags extracranial disease by approximately 15 to 20 years; stable lesions are more common in the ICA, while more dynamic, progressive lesions are found in the MCA, ACA, and posterior cerebral arteries.
Importantly, intracranial atherosclerotic disease (ICAD) accounts for approximately 9 percent of strokes in white populations but 30 to 50 percent of strokes in East Asian, Black, and Hispanic populations [73], [74]. This racial disparity persists after adjustment for traditional risk factors, suggesting underlying genetic and structural contributions. The SAMMPRIS trial established medical management as superior to stenting for symptomatic ICAD with ≥70 percent stenosis [73].
6.2 Coronary Vasculature and Aorta
Coronary atherosclerosis typically initiates in the proximal segments of the epicardial arteries, with the proximal LAD showing the highest plaque prevalence — particularly within the first 40 mm — owing to its acute take-off angle and the high flow disturbance generated at the bifurcations of diagonal and septal branches. Bifurcations of the LAD with diagonals, the LCx with obtuse marginals, and the RCA with the posterior descending artery all show predilection at the lateral, low-shear walls of the side branches.
The Pathobiological Determinants of Atherosclerosis in Youth (PDAY) study confirmed that fatty streaks and raised lesions are present in the coronaries of individuals as young as 15–34 years. In the aorta, a clear gradient of susceptibility exists: the abdominal aorta is more severely affected than the thoracic aorta, with the highest concentration of plaques occurring in the distal abdominal aorta and at the aortic bifurcation.
6.3 Renal and Mesenteric Arteries
Atherosclerotic renal artery stenosis (ARAS) accounts for approximately 90 percent of renal artery stenosis cases and primarily involves the renal artery ostia and the proximal 2 cm of the main renal artery. By contrast, fibromuscular dysplasia, which accounts for the remaining ≈10 percent, typically affects the middle and distal segments or intrarenal branches. Mesenteric artery disease — involving the celiac trunk and the superior and inferior mesenteric arteries — also occurs most often at the aortic origins, where flow turbulence is greatest.
6.4 Lower Extremity and Pelvic Arteries
Peripheral arterial disease follows a predictable progression from elastic to muscular arteries. Atherosclerosis typically appears first in the suprainguinal elastic arteries (aorta and iliacs) before progressing to the infrainguinal muscular arteries (femoral, popliteal, and tibial). Pelvic vascular disease frequently involves the internal pudendal artery (IPA), where significant stenosis or occlusion has been documented in approximately 54 percent of men screened for coronary artery disease — an extraordinary prevalence reflecting the small caliber and shared risk-factor exposure of these vessels. The distal branches of the IPA, including the cavernosal arteries, are uniquely susceptible because of their small diameter (0.5–1 mm), as discussed in Section 9.
7. Differentiation of Large-Artery Atherosclerosis and Cerebral Small Vessel Disease
The distinction between classic atherosclerosis and cerebral small vessel disease (cSVD) is fundamental to understanding the divergent mechanisms of vascular injury across the human body.
7.1 Why Penetrating Arterioles Do Not Develop Classic Atherosclerosis
The penetrating arterioles (40–200 μm in diameter) that supply the deep brain — basal ganglia, thalamus, internal capsule, and periventricular white matter — do not exhibit the eccentric, lipid-rich plaques typical of large-vessel atherosclerosis. Three structural and biological factors explain this divergence:
First, structural simplicity: these vessels lack the multi-layered lamellar structure and the well-defined internal and external elastic laminae that support classic plaque architecture. Second, blood–brain barrier specialization: the endothelial cells of these vessels are specialized components of the neurovascular unit, with tight junctions formed by claudin-5, occludin, and ZO-1, supported by pericytes, astrocyte end-feet, and a unique metabolic environment that differs fundamentally from systemic vessels. Third, the absence of vasa vasorum: these vessels rely entirely on luminal and external diffusion and do not possess the intrinsic microvascular network that can be co-opted for plaque nourishment in larger arteries.
7.2 The Spectrum of Small Vessel Pathology
Instead of classic atherosclerosis, penetrating arterioles develop a distinct set of pathologies collectively termed cerebral small vessel disease. Lipohyalinosis, originally characterized by C. Miller Fisher as “segmental arteriolar wall disorganization,” involves accumulation of waxy, glassy lipid and protein aggregates within the vessel wall, fibrinoid necrosis of medial smooth-muscle cells, and luminal narrowing. It is driven primarily by chronic hypertension and is the principal substrate of lacunar infarcts in the lenticulostriate, thalamoperforating, and pontine penetrating arterioles.
Hyperplastic arteriolosclerosis involves concentric “onion-skin” thickening of the wall due to smooth-muscle proliferation and basement-membrane duplication and is more characteristic of malignant or accelerated hypertension. Microatheroma refers to occlusive lesions in larger penetrating vessels (200–800 μm); these share some features with atherosclerosis (foam cells, lipid retention) but typically occur at the proximal origin of the perforator and reflect the spillover of large-artery disease into branches.
Cerebral amyloid angiopathy (CAA) involves progressive deposition of β-amyloid (Aβ) — predominantly Aβ40 and to a lesser extent Aβ42 — in the media and adventitia of cortical and leptomeningeal arterioles (typically <2 mm in caliber). CAA is independent of hypertension and is a major contributor to lobar microbleeds, superficial siderosis, and convexity subarachnoid hemorrhage [23], [24]. Pathologically advanced cSVD frequently shows a transition from endothelial dysfunction to BBB disruption: failure of the BBB allows toxic serum components — fibrinogen, IgG, complement — to extravasate into the brain, triggering perivascular inflammation, “forced dilatation,” and connective-tissue accumulation that leaves downstream vessels vulnerable to high-pressure damage.
8. Early-Life Evidence and Longitudinal Risk Trajectories
8.1 Napoli/FELIC: Fetal Fatty Streaks
The earliest documented atherosclerotic lesion in humans is fetal. Napoli and colleagues, in the FELIC (Fate of Early Lesions in Children) study, demonstrated that fatty streaks form in the aortic intima of fetuses, with intimal accumulation of LDL and its oxidation preceding monocyte recruitment. Fetal fatty-streak formation was greatly enhanced by maternal hypercholesterolemia during pregnancy, suggesting that the maternal lipid environment programs offspring vascular vulnerability decades before clinical disease [52].
These observations transformed the conception of atherosclerosis from a disease of mid-life to a life-course disease initiated in utero, with the prenatal environment establishing the trajectory of subsequent intimal LDL accumulation.
8.2 PDAY and the Bogalusa Heart Study
The PDAY study performed standardized autopsies on more than 3,000 individuals aged 15–34 who died of trauma, cataloguing the prevalence and extent of fatty streaks and raised lesions in the coronary arteries and aorta. Fatty streaks were present in essentially all aortas and in the coronaries of a majority by the late twenties; raised lesions appeared in approximately 20 percent of men aged 30–34. The study developed the PDAY risk score, which demonstrated that smoking, non-HDL cholesterol, and hypertension in youth are strong predictors of advanced calcification decades later.
The Bogalusa Heart Study extended these observations to a longitudinal community-based cohort, demonstrating that risk factors measured at ages 5–17 predict subclinical morbidity in adulthood. Berenson and colleagues, in autopsy data from 204 youths aged 2–39 who died of trauma, documented aortic fatty streaks in approximately half of children aged 2–15, rising to nearly 100 percent by age 21; coronary fatty streaks in 8 percent of children aged 2–15 and 69 percent of those aged 26–39; and coronary raised lesions in 3 percent of those aged 6–15 and 30 percent of those aged 26–39. The number of cardiovascular risk factors directly correlated with the extent of both fatty streaks and fibrous plaques [53].
8.3 The CARDIA Study
The Coronary Artery Risk Development in Young Adults (CARDIA) study followed 5,115 participants from ages 18–30 across more than 35 years of follow-up. Several findings have shaped contemporary preventive cardiology. First, sustained exposure to even modestly elevated LDL-C and blood pressure during the twenties and thirties contributes to ASCVD risk independently of risk levels later in life. Second, by Year 25 (mean age 50), approximately 27.7 percent of participants had detectable coronary artery calcium and approximately 53 percent had abdominal aortic calcium. Third, participants who adopted healthy habits — diet, exercise, smoking cessation, weight maintenance — during young adulthood had significantly less subclinical disease in middle age, with absolute reductions in event risk substantially larger than what mid-life intervention can achieve.
8.4 Pediatric and Adolescent Atherosclerosis: Familial Hypercholesterolemia
Heterozygous familial hypercholesterolemia (HeFH; prevalence ≈1:250) presents with LDL-C of 190–400 mg/dL from birth, the result of pathogenic variants in LDLR, APOB, or rarely PCSK9. Untreated, HeFH carries an approximately 50 percent CHD risk by age 50 in men. CIMT is significantly elevated in HeFH children by age 8–10. The landmark trial by Wiegman and colleagues demonstrated that statin initiation between ages 8 and 18 normalized CIMT progression compared with peers [54]. The 20-year follow-up of that cohort (Luirink and colleagues) showed that early statin treatment reduced MI risk by approximately 75 percent compared with untreated parents — among the strongest demonstrations in any field of medicine that early, sustained intervention can fundamentally alter a genetically determined disease trajectory [55].
9. Functional Impact of Subclinical Vascular Disease in Young and Middle-Aged Adults
Subclinical atherosclerosis is often described as “silent,” yet rigorous research demonstrates measurable functional deficits well before traditional clinical thresholds are reached.
9.1 Cognitive Function and Neurovascular Decay
In the CARDIA cohort, higher levels of CAC and abdominal aortic calcium at ages 43–55 were significantly associated with worse scores on tests of psychomotor speed (Digit Symbol Substitution Test), sustained attention, and verbal memory. This relationship persists after adjustment for age, sex, education, and traditional risk factors, suggesting that advanced calcified lesions in mid-life reflect a lifetime of vascular stress that also affects the brain microvasculature and white-matter integrity. Intelligence at age 19 has been found to inversely correlate with carotid plaque status at age 60, an association likely mediated by the long-term influence of cognitive ability on socioeconomic status, healthcare access, and adherence to healthy lifestyles.
9.2 Aerobic Capacity and Exercise Tolerance
The relationship between physical activity and subclinical disease is bidirectional. High cardiorespiratory fitness in young adulthood protects against the development of CAC and increased CIMT 15 to 25 years later. Conversely, the presence of subclinical atherosclerosis subtly impairs exercise tolerance: subclinical disease reduces vascular reserve — the ability of arteries to dilate and increase flow during peak exertion — contributing to earlier fatigue and reduced V̇O₂ peak. Individuals with low cardiorespiratory fitness are two to three times more likely to die prematurely from ASCVD even when matched for traditional risk factors.
9.3 Sleep, Fatigue, and Mood
Extreme sleep durations (<6 or >8 hours) and poor subjective sleep quality are associated with increased CAC prevalence and higher pulse-wave velocity. Sleep-duration irregularity — variation greater than 120 minutes across a week — is linked to a 33 percent higher prevalence of high CAC burden. Circadian misalignment drives chronic low-grade inflammation and sympathetic nervous system activation, predisposing individuals to subclinical atherosclerosis. Although direct causal links to mood disorders are still emerging, the combination of vascular-driven fatigue, impaired sleep, and chronic inflammation contributes to reduced quality of life.
9.4 Erectile Dysfunction as a Sentinel Event: The Artery-Size Hypothesis
Erectile dysfunction (ED) is often the first clinical manifestation of systemic vascular disease. The internal pudendal artery (1–2 mm) and its cavernosal branches (0.5–1 mm) are markedly smaller than the proximal coronary arteries (3–4 mm), the internal carotid (5–7 mm), or the femoral artery (6–8 mm). According to the artery-size hypothesis articulated by Montorsi and colleagues, equivalent atherosclerotic plaque burden produces hemodynamically significant stenosis in small vessels first, so the cavernosal circulation reaches the threshold for symptomatic compromise years before the coronary circulation does [80].
Men with vascular ED have a markedly higher prevalence of subclinical CAD; the COBRA trial reported that vasculogenic ED preceded the symptomatic onset of CAD by a mean of approximately 3 years (range 2 to 5 years) [80]. This temporal relationship makes ED a clinically actionable sentinel: every man presenting with vasculogenic ED warrants formal cardiovascular risk assessment, often including CAC scoring, lipid profiling with apoB and Lp(a), and consideration of antiplatelet and lipid-lowering therapy.
9.5 Renal Function and Renal Reserve
In young, non-hypertensive adults, renal function (estimated glomerular filtration rate, eGFR) is independently associated with arterial stiffness measured by brachial-ankle pulse wave velocity (baPWV). Mediation analysis indicates that eGFR mediates the relationship between both systolic and diastolic blood pressure and subclinical atherosclerosis, particularly in males, suggesting that elevated blood pressure influences cardiovascular health partly through early reduction in renal reserve, with secondary endocrine and oxidative changes that accelerate atherosclerotic progression.
10. Sex-Specific Differences in Subclinical Atherosclerosis
Cardiovascular disease has historically been studied in male-predominant cohorts, and the recognition of distinct female phenotypes has lagged the corresponding biology by decades. Several mechanisms produce sex-specific differences in subclinical disease.
10.1 Coronary Microvascular Dysfunction
Women are disproportionately affected by coronary microvascular dysfunction (CMD), which is the dominant mechanism of ischemia in 50–60 percent of women with angina and non-obstructive coronary arteries. The Women’s Ischemia Syndrome Evaluation (WISE) study established CMD as a major sex-specific subclinical phenotype with prognostic implications equivalent to obstructive CAD [38].
10.2 INOCA and MINOCA
Ischemia with non-obstructive coronary arteries (INOCA) refers to symptomatic ischemia in the presence of <50 percent coronary stenosis; approximately 70 percent of patients are women. MI with non-obstructive coronary arteries (MINOCA) refers to MI with <50 percent stenosis, accounts for 5–15 percent of all MIs, and is two to three times more common in women than men. Mechanisms include microvascular dysfunction, plaque erosion (rather than rupture), epicardial vasospasm, and spontaneous coronary artery dissection. Diagnosis requires invasive coronary functional testing — acetylcholine provocation, adenosine-induced flow reserve, and intravascular imaging — modalities still inconsistently available outside specialized centers [39].
10.3 Pregnancy as a Vascular Stress Test
Adverse pregnancy outcomes — preeclampsia, gestational hypertension, gestational diabetes, preterm delivery — confer a 2- to 4-fold lifetime increase in cardiovascular events. Pregnancy is now recognized as a “physiological stress test” that exposes latent vascular and metabolic dysfunction; preeclampsia in particular is associated with elevated CIMT, increased PWV, and altered endothelial function years to decades after the index pregnancy [40]. A 2020 American Heart Association scientific statement recommends incorporating pregnancy history into routine cardiovascular risk assessment for women.
10.4 Menopause Transition and Accelerated Subclinical Progression
The Study of Women’s Health Across the Nation (SWAN) demonstrated accelerated CIMT progression and arterial stiffening across the late perimenopause and early postmenopause, with median CIMT progression approximately doubling in the year before to year after the final menstrual period. Estrogen withdrawal removes its tonic effects on lipid metabolism, endothelial function, and vascular smooth-muscle phenotype. The SWAN findings have driven the recognition that the menopause transition is a vulnerable window for primary prevention rather than a static post-event endpoint [41].
10.5 Spontaneous Coronary Artery Dissection
Spontaneous coronary artery dissection (SCAD) is responsible for a disproportionate share of acute coronary syndromes in young women: more than 90 percent of SCAD cases occur in women, particularly in the peripartum period and in those aged 40–55. Approximately half of SCAD patients have coexisting fibromuscular dysplasia. SCAD is non-atherosclerotic, but its identification has reshaped the differential diagnosis of MI in young women [42].
| Phenotype | Female-predominant? | Mechanism | Detection |
| INOCA | Yes (~70%) | Microvascular dysfunction; vasospasm | Acetylcholine provocation; CFR |
| MINOCA | Yes (2–3×) | Plaque erosion; SCAD; vasospasm | OCT; IVUS |
| SCAD | Yes (>90%) | Intramural hematoma in coronary wall | Coronary angio + OCT |
| Preeclampsia legacy | Female-only | Endothelial sensitization; HTN risk | Lifetime BP; CIMT |
| Menopause-accelerated CIMT | Yes | Estrogen withdrawal | Serial CIMT; PWV |
Table 3. Female-predominant subclinical and clinical phenotypes of cardiovascular disease. CFR, coronary flow reserve; OCT, optical coherence tomography; IVUS, intravascular ultrasound; SCAD, spontaneous coronary artery dissection; CIMT, carotid intima-media thickness; PWV, pulse wave velocity.
11. The Vascular–Neurodegenerative Interface: Atherosclerosis and Alzheimer’s Disease
The historical binary distinction between vascular dementia and Alzheimer’s disease (AD) is increasingly untenable. Vascular factors — both large-vessel atherosclerosis and small vessel disease — are now recognized as central contributors to the development and trajectory of AD pathology, and a substantial fraction of clinically diagnosed AD has mixed vascular and neurodegenerative pathology at autopsy.
11.1 Oligemia and Amyloid Clearance
Severe atherosclerosis of the circle of Willis is significantly more prevalent in AD brains than in age-matched controls. Reduced cerebral perfusion — “oligemia” rather than frank ischemia — facilitates accumulation of β-amyloid (Aβ) by both increasing its production and impairing its clearance through perivascular and glymphatic pathways. Aβ itself is vasoactive, producing constriction of cerebral arteries and further aggravating the oligemic state in a self-reinforcing cycle of neurovascular decay.
11.2 Blood–Brain Barrier and Hippocampal Damage
Recent work has documented that systemic atherosclerosis is associated with amyloid and tau pathology mediated by BBB dysfunction in the hippocampus [23]. Vascular damage produces endothelial and smooth-muscle apoptosis, exacerbating cerebral amyloid angiopathy and promoting perivascular tau accumulation. Macrophages within atherosclerotic plaques can process platelet-derived amyloid precursor protein into Aβ40 and Aβ42, providing a direct biological link between systemic ApoB-driven atherosclerosis and neurodegenerative disease.
12. Inflammation in Subclinical Atherogenesis
12.1 hs-CRP and Residual Inflammatory Risk
High-sensitivity C-reactive protein (hs-CRP) integrates upstream interleukin-6 signaling and has been validated as an independent predictor of vascular events in JUPITER and multiple subsequent trials [28]. In statin-treated patients, “residual inflammatory risk” — defined as hs-CRP ≥2 mg/L despite LDL <70 mg/dL — remains a powerful predictor of recurrent events; in many secondary-prevention cohorts, residual inflammatory risk now exceeds residual cholesterol risk in magnitude [29].
12.2 The IL-1β / IL-6 Axis: CANTOS
The Canakinumab Anti-Inflammatory Thrombosis Outcome Study (CANTOS) provided randomized evidence that targeting inflammation in the absence of any LDL-lowering effect can reduce cardiovascular events. Canakinumab — a monoclonal antibody against IL-1β — reduced the primary major adverse cardiovascular events (MACE) endpoint by approximately 15 percent at the 150 mg dose without lowering LDL-C, with the greatest benefit in those with the largest reduction in IL-6 [30]. The trial supports the conclusion that inflammatory signaling contributes to recurrent events through mechanisms not fully captured by LDL-C reduction alone, and it identifies the NLRP3 inflammasome → IL-1β → IL-6 → CRP axis as a clinically tractable therapeutic target. Atherosclerosis is now best framed as a disease driven by parallel and partially independent processes — apoB-particle retention and innate immune activation — both of which can be addressed for optimal event reduction.
12.3 Clonal Hematopoiesis of Indeterminate Potential
Clonal hematopoiesis of indeterminate potential (CHIP) refers to the presence of acquired somatic mutations in hematopoietic stem cells — most commonly in DNMT3A, TET2, ASXL1, and JAK2 — without overt hematologic malignancy. CHIP is detectable in fewer than 1 percent of young adults but in approximately 10 percent of individuals over age 70. Jaiswal and colleagues demonstrated that CHIP carriers have approximately 2-fold increased risk of coronary heart disease and earlier MI (by 5–10 years) [31]. Mechanistically, mutant myeloid cells secrete excess IL-1β and IL-6, accelerating vascular inflammation; consistent with this, several studies have reported greater subclinical atherosclerotic burden or progression in CHIP carriers, although effect sizes vary by mutation, cohort, and imaging endpoint. CHIP represents a major emerging axis of cardiovascular risk that is not detected by traditional risk-factor measurement.
12.4 Neutrophil-to-Lymphocyte Ratio
The neutrophil-to-lymphocyte ratio (NLR), readily computable from any complete blood count, is an inexpensive marker of systemic inflammation. Reviews and meta-analyses associate higher NLR with increased cardiovascular risk and accelerated subclinical atherosclerosis, but cutoffs vary by population and clinical setting; the commonly cited NLR threshold of >3.0 is best presented as a research-context cutoff rather than as a guideline-endorsed diagnostic threshold [32].
13. Lipoprotein(a) as an Independent Causal Risk Factor
13.1 Genetic and Mendelian Evidence
Lipoprotein(a) [Lp(a)] is predominantly genetically determined by variation at the LPA locus on chromosome 6q26-q27. The Copenhagen General Population Study demonstrated a stepwise dose-response relationship between LPA kringle IV-2 copy-number variation and myocardial infarction risk; Mendelian-randomization studies have confirmed causality with effect sizes greater per unit than those of LDL-C [20], [21], [27].
13.2 Mechanisms of Pathogenicity
Lp(a) consists of an LDL-like particle (one apoB-100 molecule, cholesterol, phospholipids) covalently linked via a single disulfide bond between Cys4326 of apoB and Cys4057 of apolipoprotein(a) [apo(a)]. Apo(a) is structurally homologous to plasminogen, possessing 10 kringle IV repeats and a single kringle V domain, but lacks proteolytic activity. Multiple mechanisms of pathogenicity converge:
First, Lp(a) is the principal carrier of oxidized phospholipids on apoB lipoproteins; approximately 85 percent of plasma OxPL associated with apoB are bound to Lp(a). OxPL activate endothelial cells, monocytes, and platelets [22]. Second, the kringle IV-10 lysine-binding site of apo(a) competes with plasminogen for fibrin, impairing fibrinolysis and rendering Lp(a) prothrombotic [23]. Third, Lp(a) induces IL-6, IL-8, and MCP-1 expression in vascular cells, contributing to the pro-inflammatory phenotype. Fourth, Lp(a) is associated with calcific aortic valve stenosis, with Mendelian-randomization data establishing a causal link [24].
13.3 Prevalence and Clinical Implications
Approximately 20 percent of the global population has Lp(a) >50 mg/dL (>125 nmol/L), the threshold above which cardiovascular risk is materially elevated; approximately 1 in 5 individuals with premature MI have elevated Lp(a) [25], [26]. Distribution differs by ancestry: highest in West Africans, intermediate in Europeans, and lowest in East Asians.
Elevated Lp(a) should be treated as an independent causal risk factor that can coexist with absent, mild, or advanced coronary artery calcium. In MESA and the Dallas Heart Study, elevated Lp(a) and CAC were each independently associated with ASCVD risk, and participants with both elevated Lp(a) and CAC ≥100 had the highest observed risk [87]. Once-in-a-lifetime Lp(a) measurement is now supported by contemporary EAS/ESC guidance and by the 2024 National Lipid Association focused update on Lp(a) in clinical practice [25], [86]. Subclinical disease evaluation should include consideration of Lp(a)-driven phenotypes when CAC, plaque burden, aortic valve calcification, or events occur out of proportion to traditional risk factors. Emerging therapeutics — antisense oligonucleotides (pelacarsen) and siRNA agents targeting LPA mRNA — can lower Lp(a) by 80–98 percent and are currently in phase 3 cardiovascular outcome trials.
14. Microvascular Contributions and Capillary Rarefaction
14.1 Capillary Density Loss in Hypertension
Capillary rarefaction — a reduction in the number of perfused capillaries per unit tissue volume — is a hallmark of essential hypertension and precedes the development of fixed BP elevation. Antonios and colleagues demonstrated approximately 14 percent reduction in dorsal-finger capillary density in hypertensives versus normotensives [56]. Rarefaction increases peripheral vascular resistance and contributes to organ dysfunction across the kidneys, retina, and heart.
14.2 Coronary Microvascular Dysfunction
Coronary microvascular dysfunction (CMD) affects approximately half of women with chest pain and a quarter of men. Diagnosis is based on PET-derived coronary flow reserve <2.0 or invasively measured index of microcirculatory resistance (IMR) >25. The long-term prognosis is sobering: even in the absence of obstructive epicardial disease, CMD doubles the risk of MACE. The ISCHEMIA trial substudy and the WISE-CVD continuation studies have established CMD as a clinically important entity [37], [38].
14.3 Retinal Microvasculature as a Window
Retinal arteriolar narrowing — quantified by lower arteriole-to-venule ratio on fundoscopy or by optical coherence tomography angiography — predicts cardiovascular events independently of traditional risk factors. The retina is the only vascular bed directly visualizable in vivo and provides a non-invasive read-out of systemic microvascular health [57].
15. Mechanisms of Asymptomatic Progression: Glagov Remodeling and Vulnerable Plaque
15.1 Outward Remodeling and the Glagov Phenomenon
Glagov and colleagues, in 1987, demonstrated that human coronary arteries undergo a two-stage remodeling process in response to plaque accumulation. In the compensatory phase, while plaque area remains less than approximately 40 percent of the area bounded by the internal elastic lamina, the total vessel area increases such that the lumen area remains approximately constant or even slightly increases [77]. This outward remodeling allows substantial plaque burden to coexist with no restriction of resting blood flow — and thus no symptoms and no abnormality on standard luminography. Only in the encroachment phase, when plaque area exceeds ≈40 percent of the IEL area, does the vessel become unable to expand further, and the lumen begins to narrow rapidly. This biology explains why coronary angiography systematically underestimates plaque burden and why a single negative coronary angiogram does not exclude clinically meaningful subclinical disease.
15.2 Flow Reserve and Collateralization
The cardiovascular system possesses substantial functional reserve. In the coronary and renal beds, resting blood flow is typically maintained until stenosis exceeds 60–70 percent of luminal diameter; in skeletal-muscle beds, flow reserves are even higher. Slow progression of subclinical disease often allows the development of collateral circulation — alternative vascular pathways that bypass obstructive lesions — further masking the primary disease and contributing to the asymptomatic course.
15.3 The Vulnerable Plaque Concept
Not all plaques are equally dangerous. The Virmani–Stary classification defined the “thin-cap fibroatheroma” (TCFA) as a plaque with a fibrous cap thinner than 65 μm overlying a large lipid-rich necrotic core (>10 percent of plaque volume), with macrophage infiltration of the cap, intraplaque hemorrhage, and positive (outward) remodeling [81], [82]. The PROSPECT trial used three-vessel intravascular ultrasound with virtual histology to characterize 697 patients with acute coronary syndromes and showed that culprit lesions of subsequent events were predominantly TCFAs with plaque burden ≥70 percent and minimum lumen area ≤4.0 mm² [72].
| Feature | Threshold | Hazard ratio for MACE |
| Fibrous cap thickness | <65 μm (TCFA) | ≈5–7× |
| Necrotic core volume | >10% of plaque volume | ≈2–3× |
| Plaque burden | ≥70% cross-section | ≈5× (PROSPECT) |
| Minimum lumen area (IVUS) | ≤4.0 mm² | ≈3× |
| Positive remodeling index | ≥1.10 | ≈2.5× |
| Low-attenuation plaque (CCTA) | <30 HU | ≈2.5–3× |
| Napkin-ring sign (CCTA) | Qualitative | ≈5× |
Table 4. Vulnerable-plaque features and approximate hazard ratios for major adverse cardiovascular events. Ranges synthesized from PROSPECT, ICONIC, SCOT-HEART, and CRISP-CT data [63], [64], [65], [67], [72].
16. Pulse Wave Velocity, Arterial Stiffness, and Vascular Calcification
16.1 Two Distinct Calcification Phenotypes
Vascular calcification is not monolithic. Two distinct phenotypes coexist with different mechanisms, prognoses, and therapeutic implications. Intimal calcification occurs within lipid-rich atherosclerotic plaques and is the substrate quantified by the Agatston-method coronary artery calcium score; it is apoB-driven and predicts events. Medial calcification, classically described by Mönckeberg, occurs within the tunica media independently of lipids and is driven by osteogenic transdifferentiation of vascular smooth-muscle cells; it is most prominent in chronic kidney disease, type 2 diabetes, and aging [58].
16.2 Elastin Fragmentation
Aortic elastin has a half-life of approximately 70 years and is essentially non-renewable in the adult vasculature. Cyclic mechanical stress, MMP-2/MMP-9 activity, and elastolytic enzymes such as cathepsins S and K cause elastin fragmentation that directly increases pulse wave velocity. Loss of elastin recoil shifts mechanical load to collagen — a fiber 100 to 1000 times stiffer than elastin — producing the progressive aortic stiffening characteristic of vascular aging.
16.3 Calcium-Phosphate Crystallization
In CKD, hyperphosphatemia drives VSMC apoptosis and matrix-vesicle release, which nucleate hydroxyapatite crystals. Pyrophosphate, fetuin-A, and matrix Gla protein are physiological inhibitors that decline with age and CKD. Klotho — a transmembrane and circulating protein expressed in kidney that serves as co-receptor for FGF23 — declines with age, and klotho deficiency directly accelerates vascular calcification. FGF23 itself is independently associated with left ventricular hypertrophy, vascular calcification, and cardiovascular mortality [58], [59].
16.4 Pulse Wave Velocity Cutoffs
Pulse wave velocity, the gold-standard non-invasive measure of arterial stiffness, has well-established prognostic thresholds. Carotid–femoral PWV (cfPWV) is the gold standard; the European Society of Hypertension/European Society of Cardiology consensus document established >10 m/s as the threshold for elevated cardiovascular risk after correction for the actual travel path of the pulse wave [60]. Brachial–ankle PWV (baPWV), used predominantly in East Asian populations, has a commonly applied threshold of >14 m/s for elevated risk and >18 m/s for severe arterial stiffening [61].
16.5 Augmentation Index and CAVI
Augmentation index (AIx@75), derived from pulse wave analysis, reflects wave reflection from the periphery and is elevated in arterial stiffening; thresholds >25 percent are associated with elevated risk [60]. The cardio-ankle vascular index (CAVI) is a BP-independent measure of arterial stiffness, with values >9 indicating elevated risk [62].
17. Detection Modalities for Subclinical Vascular Disease
17.1 Coronary Artery Calcium Scoring
Non-contrast CT scanning of the heart with computation of the Agatston score remains the most widely validated single test for subclinical coronary disease. CAC of zero in asymptomatic adults confers a very low 10-year MACE risk, and CAC is incorporated as a risk-decision tool in the 2018 AHA/ACC cholesterol guidelines and the 2019 ESC/EAS dyslipidemia guidelines. The radiation dose is approximately 1 mSv, comparable to natural background exposure for several months.
17.2 Coronary CT Angiography and High-Risk Plaque Features
Coronary CT angiography (CCTA) provides whole-vessel morphology and the ability to identify high-risk plaque features that are not captured by Agatston scoring alone. These features include low-attenuation plaque (<30 HU within plaque, indicating lipid-rich necrotic core; HR for MACE ≈2.5–3.0); positive remodeling (remodeling index ≥1.10); spotty calcification; and the napkin-ring sign (central low attenuation surrounded by a rim of higher attenuation; HR ≈5) [63], [64].
The five-year SCOT-HEART analysis demonstrated that CCTA-guided management reduced fatal and non-fatal MI by 41 percent compared with standard care in symptomatic patients [65]. The PROMISE and CONFIRM2 registries have provided further prognostic validation.
17.3 Pericoronary Fat Attenuation Index
Pericoronary fat attenuation index (FAI), introduced by Antonopoulos and colleagues, quantifies CT attenuation of pericoronary adipose tissue within a defined radial zone. Inflamed coronary segments shift the local adipose attenuation toward less negative HU values (less lipid, more aqueous), reflecting paracrine inflammatory signaling between coronary plaque and surrounding fat [66]. The CRISP-CT study demonstrated that elevated perivascular FAI predicts cardiac mortality independently of plaque burden, with the strongest signal observed for high FAI around the proximal right coronary artery [67]. FAI-derived metrics are now incorporated into commercial post-processing platforms; U.S. FDA clearance has been reported for some products in this family (e.g., CaRi-Plaque), while CaRi-Heart/FAI-Score has regulatory clearance in selected non-U.S. markets and remains in evolving regulatory status in the United States, so specific labeling claims should be verified by product and date.
17.4 AI-Quantitative CCTA
AI-enhanced and deep-learning quantitative coronary CT angiography (AI-QCT) — exemplified by the Cleerly and HeartFlow Plaque Analysis platforms — automates segmentation of total plaque volume; calcified, non-calcified, and low-attenuation components; remodeling indices; and pericoronary fat attenuation. A 2022 international multicenter study by Lin and colleagues validated a deep-learning CCTA pipeline against expert readers and demonstrated favorable reproducibility and prognostic performance for plaque, stenosis, and risk prediction [88]. AI-QCT methods are increasingly used clinically, but specific platform claims should be matched to platform-specific validation data.
17.5 Vessel Wall MRI
High-resolution 3-Tesla vessel-wall MRI with black-blood sequences (DANTE-prepared T1, SPACE, MERGE) directly images the arterial wall, allowing differentiation of intracranial atherosclerosis (eccentric, peripheral enhancement) from vasculitis (concentric enhancement) and dissection (intramural hematoma). VW-MRI detects plaque before luminal narrowing and is particularly valuable in evaluating intracranial atherosclerotic disease [69].
17.6 Optical Coherence Tomography
Intravascular optical coherence tomography (OCT) provides 10-μm-resolution cross-sectional imaging of coronary plaques, capable of measuring fibrous cap thickness directly and identifying microcalcifications, plaque erosion, and intracoronary thrombus. OCT is the imaging modality of choice when characterizing plaque morphology in MINOCA and SCAD.
17.7 Ultrasound Microvascular Imaging
Contrast-enhanced ultrasound and super-resolution ultrasound localization microscopy (ULM) image vasa vasorum neovascularization within carotid plaques and detect microvascular rarefaction at micron-scale resolution, providing a functional read-out of plaque inflammation and tissue microcirculation [70].
17.8 Carotid Intima-Media Thickness, Ankle-Brachial Index, and Renal Doppler
Carotid intima-media thickness >0.9 mm (and focal IMT >1.5 mm defining plaque) on high-resolution B-mode ultrasound predicts cardiovascular events and is widely used in pediatric and FH research. Ankle-brachial index <0.9 indicates significant peripheral arterial disease and confers elevated cardiovascular risk irrespective of symptoms. Renal Doppler with peak systolic velocity >180–200 cm/s at the renal ostium and renal-aortic ratio >3.5 is the screening test of choice for renal artery stenosis.
| Vascular bed | Preferred modality | Key parameter / threshold |
| Coronary | Non-contrast CT (Agatston) | CAC score >0 indicates subclinical disease |
| Coronary | CCTA | LAP, NRS, PR; FAI; AI-QCT plaque volumes |
| Carotid | Ultrasound | CIMT >0.9 mm; plaque defined as focal IMT >1.5 mm |
| Lower limb | Ankle-brachial index | ABI <0.9 indicates significant PAD |
| Brain (large) | Vessel-wall MRI | Eccentric wall thickening; contrast enhancement |
| Systemic | Pulse wave velocity | cfPWV >10 m/s; baPWV >14 m/s |
| Renal | Duplex ultrasound / MRA | PSV >180–200 cm/s; renoaortic ratio >3.5 |
| Endothelial | Brachial FMD; EndoPAT RHI | FMD <7%; RHI <1.67 |
Table 5. Preferred imaging and physiological modalities for detection of subclinical vascular disease across the arterial tree. CAC, coronary artery calcium; CCTA, coronary CT angiography; LAP, low-attenuation plaque; NRS, napkin-ring sign; PR, positive remodeling; FAI, pericoronary fat attenuation index; AI-QCT, AI-quantitative coronary CT; CIMT, carotid intima-media thickness; PWV, pulse wave velocity; FMD, flow-mediated dilation; RHI, reactive hyperemia index.
18. Genetic Factors and Polygenic Risk
18.1 The 9p21 Locus
The chromosome 9p21.3 locus was identified by genome-wide association in 2007 as the first robustly replicated CHD susceptibility region. Each risk allele confers approximately 25 percent increased CHD risk, independent of all known traditional risk factors. The locus contains the long non-coding RNA ANRIL and lies adjacent to the CDKN2A/CDKN2B tumor-suppressor genes; the proposed mechanism involves dysregulation of vascular smooth-muscle proliferation and senescence [75].
18.2 LDLR, APOB, and PCSK9 Variants
Familial hypercholesterolemia is caused by pathogenic variants in LDLR (>2,000 mutations described), APOB (R3500Q the canonical variant of familial defective apoB), or, rarely, gain-of-function variants in PCSK9. The prevalence of heterozygous FH is approximately 1 in 250 in most populations; homozygous FH is roughly 1 in 300,000. Loss-of-function variants in PCSK9 (R46L, Y142X, C679X) reduce LDL across the life course and confer 30–88 percent reductions in CHD [12].
18.3 Polygenic Risk Scores
Khera and colleagues constructed a polygenic risk score comprising 6.6 million variants and demonstrated that the top 8 percent of the population have a 3-fold increased CHD risk — an effect equivalent in magnitude to monogenic FH. Polygenic scores add incremental prognostic information beyond traditional risk factors and Mendelian variants and are increasingly being incorporated into multi-layered risk-stratification models [76].
19. Lifestyle Reversibility and Plaque Regression: The Trial Evidence
A central question for the practicing clinician is whether subclinical atherosclerosis can be reversed once established. The available imaging-based randomized and case-series evidence supports the conclusion that, with sufficient reduction in apoB-particle exposure and inflammation, both functional and structural plaque regression is achievable.
19.1 The Lifestyle Heart Trial (Ornish)
The Lifestyle Heart Trial randomized 48 patients with angiographically documented CAD to a comprehensive lifestyle intervention or usual care. The intervention combined a whole-food plant-based (WFPB) diet (≈10 percent of calories from fat), aerobic exercise, stress management with yoga and meditation, group support, and smoking cessation. Quantitative coronary angiography at 1 year showed mean percent diameter stenosis decreased from 40.0 percent to 37.8 percent in the experimental group versus an increase from 42.7 percent to 46.1 percent in controls (between-group p<0.001) [43]. At 5-year follow-up, the experimental group showed continued regression to 37.3 percent versus control progression to 51.9 percent, with approximately 2.5-fold fewer cardiac events in the intervention group [44]. The Lifestyle Heart Trial is among the best-known randomized lifestyle interventions to demonstrate angiographic regression of coronary atherosclerosis.
19.2 The Esselstyn Case Series
Esselstyn and colleagues prospectively followed 198 consecutive self-selected patients with established CAD who adopted an oil-free WFPB diet. Of these, 177 (89.4 percent) were adherent over a mean 3.7 years of follow-up. Adherent patients experienced a 0.6 percent recurrent event rate, while non-adherent patients experienced a 62 percent event rate, and several patients showed angiographically documented coronary regression. The principal limitations are well recognized: the absence of a concurrent control group, self-selected participants, adherence-related selection effects, and reliance on chart-based outcome ascertainment. The findings are best presented as supportive and hypothesis-generating, consistent with the intensive lifestyle-trial evidence above, rather than as proof that WFPB intervention is uniquely effective [45].
19.3 STARS and the Heidelberg Trial
The St Thomas’ Atherosclerosis Regression Study (STARS) randomized 90 men with CAD to usual care, a lipid-lowering diet, or diet plus cholestyramine. Quantitative angiography at 39 months showed regression in 38 percent of the diet group, 33 percent of the diet + drug group, and only 4 percent of usual-care patients, with progression in 15 percent, 12 percent, and 46 percent respectively [46]. The Heidelberg Trial (Schuler and colleagues) demonstrated, with a multifactorial intervention combining a low-fat diet and structured exercise, reduced angiographic progression and modest regression at 1 year [47].
19.4 IVUS-Documented Statin and PCSK9-Inhibitor Regression Trials
Beginning with REVERSAL in 2004, intravascular ultrasound has provided high-resolution quantification of changes in percent atheroma volume (PAV) and total atheroma volume (TAV) in response to lipid-lowering therapy. The cumulative dataset establishes a clear dose–response: progressively lower on-treatment LDL produces progressively greater plaque regression.
| Trial | Year | Therapy | LDL achieved | Outcome |
| Lifestyle Heart | 1990, 1998 | WFPB + multimodal lifestyle | ≈95 mg/dL | Stenosis −7.9% (relative); ~2.5× fewer events |
| Esselstyn series | 1999, 2014 | Oil-free WFPB diet | Variable; intensive LDL lowering | 0.6% recurrent events in adherents over mean 3.7 yr; uncontrolled, self-selected |
| STARS | 1992 | Diet ± cholestyramine | ≈125 mg/dL | 38% regression vs. 4% usual care |
| REVERSAL | 2004 | Atorvastatin 80 mg | 79 mg/dL | PAV stable (Δ −0.4%) |
| ASTEROID | 2006 | Rosuvastatin 40 mg | 60.8 mg/dL | PAV −0.98%, TAV −6.8% |
| SATURN | 2011 | Rosuva 40 vs. atorva 80 | 62 / 70 mg/dL | PAV −1.22% / −0.99% |
| GLAGOV | 2016 | Evolocumab + statin | 36.6 mg/dL | PAV −0.95% vs. +0.05% |
| PACMAN-AMI | 2022 | Alirocumab + statin | 23.5 mg/dL | PAV −2.13%; ↑ fibrous cap thickness |
| HUYGENS | 2022 | Evolocumab + statin | 28.1 mg/dL | Fibrous cap thickness +42.7 μm |
Table 6. Imaging-verified plaque regression trials, organized by intervention intensity and on-treatment LDL-C. PAV, percent atheroma volume; TAV, total atheroma volume; WFPB, whole-food plant-based [14], [15], [16], [17], [43], [44], [45], [46], [48], [49].
REVERSAL demonstrated essentially halted progression at LDL ≈79 mg/dL with high-dose statin therapy [14]. ASTEROID was the first trial to demonstrate clear regression — PAV reduced by 0.98 percent and TAV reduced by 6.8 percent — at on-treatment LDL of 60.8 mg/dL with rosuvastatin 40 mg [15]. SATURN extended the comparison to include atorvastatin 80 mg [16]. GLAGOV randomized 968 patients on optimized statin therapy to evolocumab or placebo and demonstrated PAV regression of 0.95 percent in the evolocumab arm at on-treatment LDL of 36.6 mg/dL [17]. PACMAN-AMI, in patients post-acute MI, demonstrated PAV reduction of 2.13 percent and concurrent stabilization of plaque morphology — increased fibrous cap thickness, decreased lipid pool — at on-treatment LDL of 23.5 mg/dL with alirocumab on top of high-intensity statin [48]. HUYGENS, using OCT to measure fibrous-cap thickness directly, demonstrated an increase of 42.7 μm at on-treatment LDL of 28.1 mg/dL — converting many plaques from the TCFA to the thick-cap fibroatheroma classification [49].
The combined message of these trials is that plaque regression and stabilization have been repeatedly observed with intensive LDL-C lowering, especially when achieved LDL-C is well below 70 mg/dL; the magnitude of regression varies by baseline plaque burden, imaging modality, achieved apoB/LDL-C, and background therapy.
19.5 The Subclinical Phase as the Optimal Intervention Window
Once a vulnerable plaque has formed — with its necrotic core, thin fibrous cap, and positive remodeling — even aggressive therapy can reduce and stabilize risk but does not eliminate all residual events, even at very low achieved LDL-C [18]. Conversely, primordial prevention — preventing the first transcytosis–retention event by maintaining low apoB exposure across the life course — more closely approximates the large lifetime benefit observed in genetically lower-LDL states. The operational implication is direct: start early, lower sufficiently, and sustain risk-factor control indefinitely.
19.6 The CAC Paradox
Statin therapy has been observed to increase the volume of coronary calcium even while reducing event rates. This does not necessarily indicate harmful progression: statins can increase plaque calcium density (a marker associated with healing and stabilization) while reducing the lipid-rich, vulnerable component of plaque. Because the Agatston score weights calcium density (assigning higher scores to denser calcium), a stabilizing lesion may yield a higher Agatston score even as event risk falls. The CAC density and volume score, developed by Criqui and colleagues, addresses this paradox by demonstrating that, at any given total Agatston score, higher CAC density predicts fewer events [50], [51]. The clinical implication is that serial CAC scoring during statin therapy must be interpreted with reference to density and morphology, not the Agatston number alone.
20. Synthesis and Implications for Practice
Subclinical atherosclerosis is not a passive state of aging but an active, progressive biological syndrome with measurable consequences in early adulthood — and, increasingly, in childhood and even in utero. The body of evidence reviewed here supports several integrated conclusions.
First, atherogenesis is causally driven by apoB-containing lipoproteins. The convergence of Mendelian-randomization, familial-genetic, and randomized-trial evidence across approximately twenty independent lines of investigation establishes apoB causality with rigor unmatched in most areas of medicine. Risk is proportional to the integral of apoB-particle exposure over time. The corollary is that primordial prevention — maintaining low apoB across the life course — yields the largest possible event reduction.
Second, parallel non-lipid pathways amplify or modulate apoB-driven atherogenesis. Lp(a), inflammation (NLRP3 → IL-1β → IL-6 → CRP), clonal hematopoiesis, hypertension, glycemic dysregulation, and arterial stiffening each contribute mechanistically distinct components of risk. Comprehensive prevention addresses all of these axes, not LDL alone.
Third, the divergence between large-artery atherosclerosis and cerebral small vessel disease reflects a fundamental structural and hemodynamic threshold. Vessel-wall complexity and the presence of vasa vasorum permit lipid-core plaque formation; the simpler architecture of penetrating arterioles favors lipohyalinosis, microatheroma, and amyloid angiopathy instead. Each pathology has its own risk-factor profile and optimal therapeutic approach.
Fourth, the “clinical silence” of subclinical disease is maintained by adaptive remodeling — the Glagov phenomenon — and by physiological reserve in flow capacity. This silence is regularly punctured by subtle functional decays in cognitive processing speed, sleep regularity, exercise tolerance, and erectile function. These functional signals, properly recognized, provide a clinically actionable window for intervention years before the first overt event.
Fifth, sex-specific phenotypes — INOCA, MINOCA, SCAD, preeclampsia legacy effects, menopause-accelerated CIMT progression — have historically been under-diagnosed and require dedicated clinical pathways.
Sixth, the trial evidence — from the Lifestyle Heart Trial through the modern IVUS regression trials with PCSK9 inhibitors — supports the conclusion that subclinical atherosclerosis is, when addressed early and aggressively, partially reversible. Plaque can be stabilized, fibrous caps thickened, and event rates reduced. Whole-food plant-based dietary patterns and pharmacologic LDL-lowering both produce regression in their respective trial contexts; comprehensive lifestyle intervention complements rather than replaces appropriate pharmacologic therapy.
The life-course perspective provided by Napoli/FELIC, Bogalusa, PDAY, CARDIA, MESA, and the Tsimane natural experiment carries one practical message: the vascular integrity of late life is built on the risk exposures of youth. Early detection through CAC scoring, pulse wave velocity, vessel-wall MRI, FAI, and emerging AI-augmented imaging, combined with aggressive risk-factor management addressing apoB, Lp(a), inflammation, blood pressure, glycemia, sleep, fitness, and tobacco exposure, offers the realistic possibility of compressing cardiovascular morbidity to the very end of life. The goal of preventive cardiology is no longer the deferral of the first event by a decade; it is the elimination of clinical atherosclerotic disease as a routine cause of human suffering.
References
- Ference BA, Yoo W, Alesh I, et al. Effect of long-term exposure to lower low-density lipoprotein cholesterol beginning early in life on the risk of coronary heart disease: a Mendelian randomization analysis. J Am Coll Cardiol. 2012;60(25):2631-2639. doi:10.1016/j.jacc.2012.09.017
- Ference BA, Kastelein JJP, Catapano AL. Lipids and Lipoproteins in 2020. JAMA. 2020;324(6):595-596. doi:10.1001/jama.2020.5685
- Ference BA, Robinson JG, Brook RD, et al. Variation in PCSK9 and HMGCR and Risk of Cardiovascular Disease and Diabetes. N Engl J Med. 2016;375(22):2144-2153. doi:10.1056/NEJMoa1604304
- Ference BA, Ginsberg HN, Graham I, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2017;38(32):2459-2472. doi:10.1093/eurheartj/ehx144
- Marston NA, Giugliano RP, Melloni GEM, et al. Association of Apolipoprotein B-Containing Lipoproteins and Risk of Myocardial Infarction in Individuals With and Without Atherosclerosis: Distinguishing Between Particle Concentration, Type, and Content. JAMA Cardiol. 2022;7(3):250-256. doi:10.1001/jamacardio.2021.5083
- Sniderman AD, Thanassoulis G, Glavinovic T, et al. Apolipoprotein B Particles and Cardiovascular Disease: A Narrative Review. JAMA Cardiol. 2019;4(12):1287-1295. doi:10.1001/jamacardio.2019.3780
- Ference BA, Bhatt DL, Catapano AL, et al. Association of Genetic Variants Related to Combined Exposure to Lower Low-Density Lipoproteins and Lower Systolic Blood Pressure With Lifetime Risk of Cardiovascular Disease. JAMA. 2019;322(14):1381-1391. doi:10.1001/jama.2019.14120
- Vogel RA. The new cholesterol guidelines: finally more light than heat. J Am Coll Cardiol. 2014;64(9):920-921. doi:10.1016/j.jacc.2014.06.1168
- Domanski MJ, Tian X, Wu CO, et al. Time Course of LDL Cholesterol Exposure and Cardiovascular Disease Event Risk. J Am Coll Cardiol. 2020;76(13):1507-1516. doi:10.1016/j.jacc.2020.07.059
- Kaplan H, Thompson RC, Trumble BC, et al. Coronary atherosclerosis in indigenous South American Tsimane: a cross-sectional cohort study. Lancet. 2017;389(10080):1730-1739. doi:10.1016/S0140-6736(17)30752-3
- Pontzer H, Wood BM, Raichlen DA. Hunter-gatherers as models in public health. Obes Rev. 2018;19 Suppl 1:24-35. doi:10.1111/obr.12785
- Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354(12):1264-1272. doi:10.1056/NEJMoa054013
- Stitziel NO, Khera AV, Wang X, et al. ANGPTL3 Deficiency and Protection Against Coronary Artery Disease. J Am Coll Cardiol. 2017;69(16):2054-2063. doi:10.1016/j.jacc.2017.02.030
- Nissen SE, Tuzcu EM, Schoenhagen P, et al. Effect of intensive compared with moderate lipid-lowering therapy on progression of coronary atherosclerosis: a randomized controlled trial. JAMA. 2004;291(9):1071-1080. doi:10.1001/jama.291.9.1071
- Nissen SE, Nicholls SJ, Sipahi I, et al. Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA. 2006;295(13):1556-1565. doi:10.1001/jama.295.13.jpc60002
- Nicholls SJ, Ballantyne CM, Barter PJ, et al. Effect of two intensive statin regimens on progression of coronary disease. N Engl J Med. 2011;365(22):2078-2087. doi:10.1056/NEJMoa1110874
- Nicholls SJ, Puri R, Anderson T, et al. Effect of Evolocumab on Progression of Coronary Disease in Statin-Treated Patients: The GLAGOV Randomized Clinical Trial. JAMA. 2016;316(22):2373-2384. doi:10.1001/jama.2016.16951
- Gaba P, O’Donoghue ML, Park JG, et al. Association Between Achieved Low-Density Lipoprotein Cholesterol Levels and Long-Term Cardiovascular and Safety Outcomes: An Analysis of FOURIER-OLE. Circulation. 2023;147(16):1192-1203. doi:10.1161/CIRCULATIONAHA.122.063399
- Borén J, Chapman MJ, Krauss RM, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2020;41(24):2313-2330. doi:10.1093/eurheartj/ehz962
- Kamstrup PR, Tybjaerg-Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301(22):2331-2339. doi:10.1001/jama.2009.801
- Clarke R, Peden JF, Hopewell JC, et al. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med. 2009;361(26):2518-2528. doi:10.1056/NEJMoa0902604
- Tsimikas S. A Test in Context: Lipoprotein(a): Diagnosis, Prognosis, Controversies, and Emerging Therapies. J Am Coll Cardiol. 2017;69(6):692-711. doi:10.1016/j.jacc.2016.11.042
- Koschinsky ML, Marcovina SM. Structure-function relationships in apolipoprotein(a): insights into lipoprotein(a) assembly and pathogenicity. Curr Opin Lipidol. 2004;15(2):167-174. doi:10.1097/00041433-200404000-00009
- Thanassoulis G, Campbell CY, Owens DS, et al. Genetic associations with valvular calcification and aortic stenosis. N Engl J Med. 2013;368(6):503-512. doi:10.1056/NEJMoa1109034
- Kronenberg F, Mora S, Stroes ESG, et al. Lipoprotein(a) in atherosclerotic cardiovascular disease and aortic stenosis: a European Atherosclerosis Society consensus statement. Eur Heart J. 2022;43(39):3925-3946. doi:10.1093/eurheartj/ehac361
- Nordestgaard BG, Chapman MJ, Ray K, et al. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;31(23):2844-2853. doi:10.1093/eurheartj/ehq386
- Kamstrup PR, Benn M, Tybjaerg-Hansen A, Nordestgaard BG. Extreme lipoprotein(a) levels and risk of myocardial infarction in the general population: the Copenhagen City Heart Study. Circulation. 2008;117(2):176-184. doi:10.1161/CIRCULATIONAHA.107.715698
- Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359(21):2195-2207. doi:10.1056/NEJMoa0807646
- Ridker PM. How Common Is Residual Inflammatory Risk?. Circ Res. 2017;120(4):617-619. doi:10.1161/CIRCRESAHA.116.310527
- Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med. 2017;377(12):1119-1131. doi:10.1056/NEJMoa1707914
- Jaiswal S, Natarajan P, Silver AJ, et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N Engl J Med. 2017;377(2):111-121. doi:10.1056/NEJMoa1701719
- Bhat T, Teli S, Rijal J, et al. Neutrophil to lymphocyte ratio and cardiovascular diseases: a review. Expert Rev Cardiovasc Ther. 2013;11(1):55-59. doi:10.1586/erc.12.159
- Yeboah J, Folsom AR, Burke GL, et al. Predictive value of brachial flow-mediated dilation for incident cardiovascular events in a population-based study: the multi-ethnic study of atherosclerosis. Circulation. 2009;120(6):502-509. doi:10.1161/CIRCULATIONAHA.109.864801
- Bonetti PO, Pumper GM, Higano ST, Holmes DR Jr, Kuvin JT, Lerman A. Noninvasive identification of patients with early coronary atherosclerosis by assessment of digital reactive hyperemia. J Am Coll Cardiol. 2004;44(11):2137-2141. doi:10.1016/j.jacc.2004.08.062
- van den Berg BM, Spaan JA, Rolf TM, Vink H. Atherogenic region and diet diminish glycocalyx dimension and increase intima-to-media ratios at murine carotid artery bifurcation. Am J Physiol Heart Circ Physiol. 2006;290(2):H915-H920. doi:10.1152/ajpheart.00051.2005
- Becker BF, Chappell D, Bruegger D, Annecke T, Jacob M. Therapeutic strategies targeting the endothelial glycocalyx: acute deficits, but great potential. Cardiovasc Res. 2010;87(2):300-310. doi:10.1093/cvr/cvq137
- Taqueti VR, Di Carli MF. Coronary Microvascular Disease Pathogenic Mechanisms and Therapeutic Options: JACC State-of-the-Art Review. J Am Coll Cardiol. 2018;72(21):2625-2641. doi:10.1016/j.jacc.2018.09.042
- Shaw LJ, Bairey Merz CN, Pepine CJ, et al. Insights from the NHLBI-Sponsored Women’s Ischemia Syndrome Evaluation (WISE) Study: Part I: gender differences in traditional and novel risk factors, symptom evaluation, and gender-optimized diagnostic strategies. J Am Coll Cardiol. 2006;47(3 Suppl):S4-S20. doi:10.1016/j.jacc.2005.01.072
- Tamis-Holland JE, Jneid H, Reynolds HR, et al. Contemporary Diagnosis and Management of Patients With Myocardial Infarction in the Absence of Obstructive Coronary Artery Disease: A Scientific Statement From the American Heart Association. Circulation. 2019;139(18):e891-e908. doi:10.1161/CIR.0000000000000670
- Bellamy L, Casas JP, Hingorani AD, Williams DJ. Pre-eclampsia and risk of cardiovascular disease and cancer in later life: systematic review and meta-analysis. BMJ. 2007;335(7627):974. doi:10.1136/bmj.39335.385301.BE
- El Khoudary SR, Aggarwal B, Beckie TM, et al. Menopause Transition and Cardiovascular Disease Risk: Implications for Timing of Early Prevention: A Scientific Statement From the American Heart Association. Circulation. 2020;142(25):e506-e532. doi:10.1161/CIR.0000000000000912
- Hayes SN, Kim ESH, Saw J, et al. Spontaneous Coronary Artery Dissection: Current State of the Science: A Scientific Statement From the American Heart Association. Circulation. 2018;137(19):e523-e557. doi:10.1161/CIR.0000000000000564
- Ornish D, Brown SE, Scherwitz LW, et al. Can lifestyle changes reverse coronary heart disease? The Lifestyle Heart Trial. Lancet. 1990;336(8708):129-133. doi:10.1016/0140-6736(90)91656-u
- Ornish D, Scherwitz LW, Billings JH, et al. Intensive lifestyle changes for reversal of coronary heart disease. JAMA. 1998;280(23):2001-2007. doi:10.1001/jama.280.23.2001
- Esselstyn CB Jr, Gendy G, Doyle J, Golubic M, Roizen MF. A way to reverse CAD?. J Fam Pract. 2014;63(7):356-364b.
- Watts GF, Lewis B, Brunt JN, et al. Effects on coronary artery disease of lipid-lowering diet, or diet plus cholestyramine, in the St Thomas’ Atherosclerosis Regression Study (STARS). Lancet. 1992;339(8793):563-569. doi:10.1016/0140-6736(92)90863-x
- Schuler G, Hambrecht R, Schlierf G, et al. Regular physical exercise and low-fat diet. Effects on progression of coronary artery disease. Circulation. 1992;86(1):1-11. doi:10.1161/01.cir.86.1.1
- Räber L, Ueki Y, Otsuka T, et al. Effect of Alirocumab Added to High-Intensity Statin Therapy on Coronary Atherosclerosis in Patients With Acute Myocardial Infarction: The PACMAN-AMI Randomized Clinical Trial. JAMA. 2022;327(18):1771-1781. doi:10.1001/jama.2022.5218
- Nicholls SJ, Kataoka Y, Nissen SE, et al. Effect of Evolocumab on Coronary Plaque Phenotype and Burden in Statin-Treated Patients Following Myocardial Infarction. JACC Cardiovasc Imaging. 2022;15(7):1308-1321. doi:10.1016/j.jcmg.2022.03.002
- Criqui MH, Denenberg JO, Ix JH, et al. Calcium density of coronary artery plaque and risk of incident cardiovascular events. JAMA. 2014;311(3):271-278. doi:10.1001/jama.2013.282535
- Henein M, Granåsen G, Wiklund U, et al. High dose and long-term statin therapy accelerate coronary artery calcification. Int J Cardiol. 2015;184:581-586. doi:10.1016/j.ijcard.2015.02.072
- Napoli C, D’Armiento FP, Mancini FP, et al. Fatty streak formation occurs in human fetal aortas and is greatly enhanced by maternal hypercholesterolemia. Intimal accumulation of low density lipoprotein and its oxidation precede monocyte recruitment into early atherosclerotic lesions. J Clin Invest. 1997;100(11):2680-2690. doi:10.1172/JCI119813
- Berenson GS, Srinivasan SR, Bao W, Newman WP 3rd, Tracy RE, Wattigney WA. Association between multiple cardiovascular risk factors and atherosclerosis in children and young adults. The Bogalusa Heart Study. N Engl J Med. 1998;338(23):1650-1656. doi:10.1056/NEJM199806043382302
- Wiegman A, Hutten BA, de Groot E, et al. Efficacy and safety of statin therapy in children with familial hypercholesterolemia: a randomized controlled trial. JAMA. 2004;292(3):331-337. doi:10.1001/jama.292.3.331
- Luirink IK, Wiegman A, Kusters DM, et al. 20-Year Follow-up of Statins in Children with Familial Hypercholesterolemia. N Engl J Med. 2019;381(16):1547-1556. doi:10.1056/NEJMoa1816454
- Antonios TF, Singer DR, Markandu ND, Mortimer PS, MacGregor GA. Rarefaction of skin capillaries in borderline essential hypertension suggests an early structural abnormality. Hypertension. 1999;34(4 Pt 1):655-658. doi:10.1161/01.hyp.34.4.655
- Wong TY, Klein R, Sharrett AR, et al. Retinal arteriolar narrowing and risk of coronary heart disease in men and women. The Atherosclerosis Risk in Communities Study. JAMA. 2002;287(9):1153-1159. doi:10.1001/jama.287.9.1153
- Demer LL, Tintut Y. Vascular calcification: pathobiology of a multifaceted disease. Circulation. 2008;117(22):2938-2948. doi:10.1161/CIRCULATIONAHA.107.743161
- Gutiérrez OM, Mannstadt M, Isakova T, et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med. 2008;359(6):584-592. doi:10.1056/NEJMoa0706130
- Van Bortel LM, Laurent S, Boutouyrie P, et al. Expert consensus document on the measurement of aortic stiffness in daily practice using carotid-femoral pulse wave velocity. J Hypertens. 2012;30(3):445-448. doi:10.1097/HJH.0b013e32834fa8b0
- Munakata M. Brachial-Ankle Pulse Wave Velocity: Background, Method, and Clinical Evidence. Pulse (Basel). 2016;3(3-4):195-204. doi:10.1159/000443740
- Shirai K, Utino J, Otsuka K, Takata M. A novel blood pressure-independent arterial wall stiffness parameter; cardio-ankle vascular index (CAVI). J Atheroscler Thromb. 2006;13(2):101-107. doi:10.5551/jat.13.101
- Motoyama S, Ito H, Sarai M, et al. Plaque Characterization by Coronary Computed Tomography Angiography and the Likelihood of Acute Coronary Events in Mid-Term Follow-Up. J Am Coll Cardiol. 2015;66(4):337-346. doi:10.1016/j.jacc.2015.05.069
- Maurovich-Horvat P, Ferencik M, Voros S, Merkely B, Hoffmann U. Comprehensive plaque assessment by coronary CT angiography. Nat Rev Cardiol. 2014;11(7):390-402. doi:10.1038/nrcardio.2014.60
- SCOT-HEART Investigators, Newby DE, Adamson PD, et al. Coronary CT Angiography and 5-Year Risk of Myocardial Infarction. N Engl J Med. 2018;379(10):924-933. doi:10.1056/NEJMoa1805971
- Antonopoulos AS, Sanna F, Sabharwal N, et al. Detecting human coronary inflammation by imaging perivascular fat. Sci Transl Med. 2017;9(398):eaal2658. doi:10.1126/scitranslmed.aal2658
- Oikonomou EK, Marwan M, Desai MY, et al. Non-invasive detection of coronary inflammation using computed tomography and prediction of residual cardiovascular risk (the CRISP CT study): a post-hoc analysis of prospective outcome data. Lancet. 2018;392(10151):929-939. doi:10.1016/S0140-6736(18)31114-0
- Min JK, Leipsic J, Pencina MJ, et al. Diagnostic accuracy of fractional flow reserve from anatomic CT angiography. JAMA. 2012;308(12):1237-1245. doi:10.1001/2012.jama.11274
- Kang N, Qiao Y, Wasserman BA. Essentials for Interpreting Intracranial Vessel Wall MRI Results: State of the Art. Radiology. 2021;300(3):492-505. doi:10.1148/radiol.2021204096
- Christensen-Jeffries K, Couture O, Dayton PA, et al. Super-resolution Ultrasound Imaging. Ultrasound Med Biol. 2020;46(4):865-891. doi:10.1016/j.ultrasmedbio.2019.11.013
- Ku DN, Giddens DP, Zarins CK, Glagov S. Pulsatile flow and atherosclerosis in the human carotid bifurcation. Positive correlation between plaque location and low oscillating shear stress. Arteriosclerosis. 1985;5(3):293-302. doi:10.1161/01.atv.5.3.293
- Stone GW, Maehara A, Lansky AJ, et al. A prospective natural-history study of coronary atherosclerosis. N Engl J Med. 2011;364(3):226-235. doi:10.1056/NEJMoa1002358
- Chimowitz MI, Lynn MJ, Derdeyn CP, et al. Stenting versus aggressive medical therapy for intracranial arterial stenosis. N Engl J Med. 2011;365(11):993-1003. doi:10.1056/NEJMoa1105335
- Wang Y, Meng R, Liu G, et al. Intracranial atherosclerotic disease. Neurobiol Dis. 2019;124:118-132. doi:10.1016/j.nbd.2018.11.008
- Helgadottir A, Thorleifsson G, Manolescu A, et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science. 2007;316(5830):1491-1493. doi:10.1126/science.1142842
- Khera AV, Chaffin M, Aragam KG, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet. 2018;50(9):1219-1224. doi:10.1038/s41588-018-0183-z
- Glagov S, Weisenberg E, Zarins CK, Stankunavicius R, Kolettis GJ. Compensatory enlargement of human atherosclerotic coronary arteries. N Engl J Med. 1987;316(22):1371-1375. doi:10.1056/NEJM198705283162204
- Wolinsky H, Glagov S. A lamellar unit of aortic medial structure and function in mammals. Circ Res. 1967;20(1):99-111. doi:10.1161/01.res.20.1.99
- Okita Y. An intimal cylinder in the descending aorta. J Thorac Cardiovasc Surg. 2015;150(3):e35-e36. doi:10.1016/j.jtcvs.2015.06.043
- Montorsi P, Ravagnani PM, Galli S, et al. Association between erectile dysfunction and coronary artery disease. Role of coronary clinical presentation and extent of coronary vessels involvement: the COBRA trial. Eur Heart J. 2006;27(22):2632-2639. doi:10.1093/eurheartj/ehl142
- Fuster V, Moreno PR, Fayad ZA, Corti R, Badimon JJ. Atherothrombosis and high-risk plaque: part I: evolving concepts. J Am Coll Cardiol. 2005;46(6):937-954. doi:10.1016/j.jacc.2005.03.074
- Virmani R, Burke AP, Farb A, Kolodgie FD. Pathology of the vulnerable plaque. J Am Coll Cardiol. 2006;47(8 Suppl):C13-C18. doi:10.1016/j.jacc.2005.10.065
- Tabas I, Williams KJ, Borén J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 2007;116(16):1832-1844. doi:10.1161/CIRCULATIONAHA.106.676890
- Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352(16):1685-1695. doi:10.1056/NEJMra043430
- Gimbrone MA Jr, García-Cardeña G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ Res. 2016;118(4):620-636. doi:10.1161/CIRCRESAHA.115.306301
- Koschinsky ML, Bajaj A, Boffa MB, et al. A focused update to the 2019 NLA scientific statement on use of lipoprotein(a) in clinical practice. J Clin Lipidol. 2024;18(3):e308-e319. doi:10.1016/j.jacl.2024.03.001
- Mehta A, Vasquez N, Ayers CR, et al. Independent Association of Lipoprotein(a) and Coronary Artery Calcification With Atherosclerotic Cardiovascular Risk. J Am Coll Cardiol. 2022;79(8):757-768. doi:10.1016/j.jacc.2021.11.058
- Lin A, Manral N, McElhinney P, et al. Deep learning-enabled coronary CT angiography for plaque and stenosis quantification and cardiac risk prediction: an international multicentre study. Lancet Digit Health. 2022;4(4):e256-e265. doi:10.1016/S2589-7500(22)00022-X