
Lifelong Cumulative Apolipoprotein B Exposure and Coronary Atherosclerosis in Chronic Endurance Athletes
A Mechanistic Hypothesis and Critical Narrative Review
Abstract
Background. Endurance training is associated with favorable cardiometabolic profiles and lower overall mortality, yet several coronary imaging studies have reported a paradoxically higher burden of subclinical coronary calcification and plaque in lifelong masters endurance athletes than in active, non-athletic controls.
Objective. To critically appraise the hypothesis that decades of high-volume endurance exercise alter the coronary arterial microenvironment in ways that may facilitate the subendothelial entry and retention of apolipoprotein B (ApoB)-containing lipoproteins, thereby potentially amplifying the atherogenic significance of cumulative ApoB exposure.
Evidence approach. This article is a narrative review and critical appraisal of the mechanistic, imaging, epidemiologic, and therapeutic literature bearing on the question. Because a reproducible search strategy, formal screening process, and prespecified protocol were not applied, it should not be interpreted as a PRISMA-compliant systematic review in its present form.
Results. The reviewed literature supports several established principles: ApoB-containing lipoproteins are causal in atherogenesis; lipoprotein retention within the arterial wall is central to plaque initiation; and endurance athletes can show a higher prevalence of coronary artery calcium and plaque than their traditional risk-factor profiles would predict. A plausible—but not yet directly proven—mechanobiological model is that repeated exposure to high exercise blood pressure, cyclic wall strain, and locally disturbed coronary flow may, at anatomically susceptible sites, alter endothelial barrier function, lipoprotein transport, or intimal matrix retention of ApoB particles. Athletes may nonetheless experience lower clinical risk at a given burden of subclinical disease because of high cardiorespiratory fitness, favorable systemic risk factors, and vascular adaptations; whether larger coronary caliber, plaque composition, or collateral function materially mediates this association remains uncertain.
Conclusions. Taken together, the evidence supports a hypothesis-generating model in which lower circulating ApoB may be a rational additive preventive strategy in selected endurance athletes—particularly those with demonstrable plaque, elevated coronary calcium, or inherited lipid risk. Athlete-specific randomized outcome trials are lacking, however, and recommendations should be framed as individualized risk-management considerations rather than outcome-proven standards.
Keywords: apolipoprotein B; endurance athletes; coronary artery calcium; coronary CT angiography; atherosclerosis; lipid management; exercise physiology
Abbreviations: ALK1, activin receptor-like kinase 1; ApoB, apolipoprotein B; ARR, absolute risk reduction; ASCVD, atherosclerotic cardiovascular disease; CAC, coronary artery calcium; CCTA, coronary CT angiography; CTT, Cholesterol Treatment Trialists’ Collaboration; eGC, endothelial glycocalyx; FAK, focal adhesion kinase; GAG, glycosaminoglycan; HR, hazard ratio; IDL, intermediate-density lipoprotein; LDL-C, low-density lipoprotein cholesterol; Lp(a), lipoprotein(a); MACE, major adverse cardiovascular events; NNT, number needed to treat; NO, nitric oxide; OR, odds ratio; RRR, relative risk reduction; SAMS, statin-associated muscle symptoms; SR-B1, scavenger receptor class B type 1; TGF-β, transforming growth factor-beta; VLDL, very-low-density lipoprotein; VSMC, vascular smooth muscle cell.
Executive Summary
The traditional cardiovascular prevention paradigm is anchored in the premise that regular aerobic physical activity improves lipid profiles, reduces blood pressure, prevents type 2 diabetes, mitigates systemic inflammation, and increases life expectancy. However, contemporary coronary imaging studies have revealed a paradoxical phenomenon: lifelong, high-volume endurance athletes—such as marathon runners, competitive cyclists, triathletes, cross-country skiers, and rowers—demonstrate a greater prevalence and volume of subclinical coronary artery calcium (CAC) and atherosclerotic plaque than active, non-athletic controls with comparably low cardiovascular risk profiles.
This narrative review and critical appraisal evaluates the hypothesis that the unique hemodynamic and mechanical microenvironment generated by decades of high-volume endurance exercise may accelerate the subendothelial retention of apolipoprotein B (ApoB)-containing lipoproteins. If this physical loading does accelerate atherogenesis, a clinical rationale may exist for maintaining lower ApoB concentrations in lifelong endurance athletes than those currently recommended for the general population.
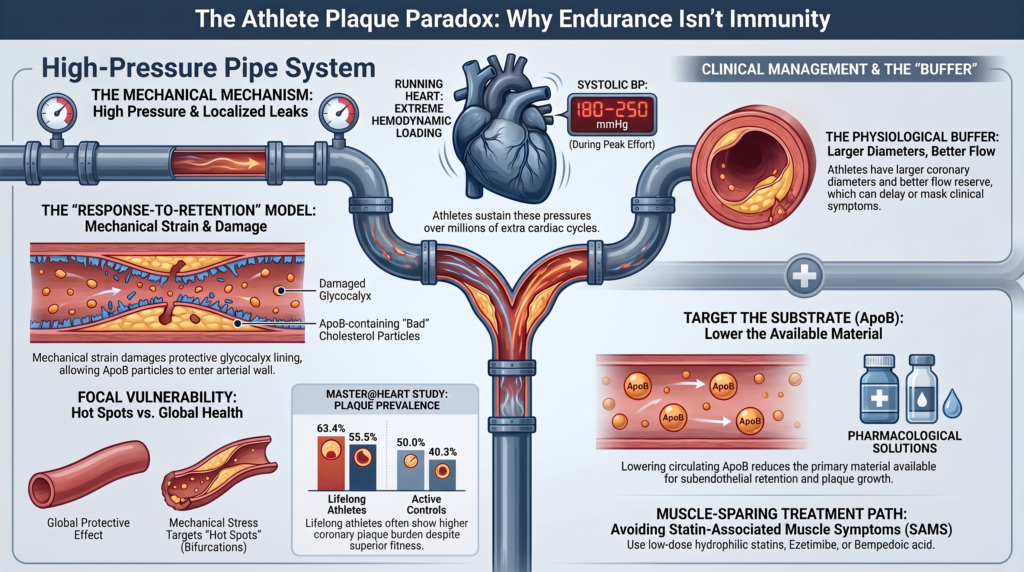
Through an integration of vascular biomechanics, the response-to-retention model of atherogenesis, clinical trials, and epidemiological outcome data, this review argues that prolonged physical training may produce biological alterations in the epicardial coronary wall that could create localized biomechanical conditions theoretically favoring ApoB retention at focal sites. Repetitive exposure to exercise-induced systolic blood pressures of roughly 180 to 220 mmHg (extending to ~250 mmHg at peak effort), tens to several hundreds of millions of additional elevated-rate cardiac cycles over decades (with greater totals theoretically possible at exceptionally high daily training volumes, though cumulative beat count has not been validated as an independent atherosclerotic exposure), and cyclic arterial wall deformation may contribute to localized remodeling or shedding of the protective endothelial glycocalyx at anatomically susceptible arterial sites.
Experimental studies suggest that cyclic mechanical strain can upregulate the synthesis of highly charged intimal proteoglycans, particularly biglycan, which act as structural anchors for ApoB retention, though whether this occurs in the athlete coronary is unknown. Endurance athletes are, in any case, substantially protected from clinical coronary events through physiological adaptations—high cardiorespiratory fitness, favorable endothelial function, and myocardial reserve—even where subclinical plaque is present in proximal coronary segments.
Consequently, lowering the absolute concentration of circulating ApoB-containing particles is a biologically plausible strategy to mitigate any biomechanically enhanced risk of subendothelial retention. It is important to state at the outset that this conclusion is hypothesis-generating: no randomized outcome trial has tested lower ApoB targets in asymptomatic athletes, and whether such targets add benefit beyond current guideline-directed lipid lowering remains unknown and should be evaluated in randomized clinical trials. The idea should be read as a reasoned extension of mechanistic and genetic evidence rather than an outcome-proven standard.
Introduction: The Athletic Atherosclerosis Hypothesis and Cumulative Lipid Exposure
The biological foundation of atherosclerotic cardiovascular disease (ASCVD) rests on the cumulative exposure of the arterial wall to apolipoprotein B (ApoB)-containing lipoproteins over time, a relationship conceptualized as the “ApoB-years” or “cholesterol-years” burden. “ApoB-years” is used here as a conceptual description of time-integrated exposure to atherogenic particles, not as a currently standardized or prospectively validated clinical metric. Circulating ApoB-containing particles—low-density lipoprotein (LDL), very-low-density lipoprotein (VLDL) remnants, intermediate-density lipoprotein (IDL), and lipoprotein(a) [Lp(a)]—are causal and generally necessary participants in the development of lipid-rich atherosclerotic plaque. When cumulative ApoB exposure is exceptionally low, the capacity of hypertension, smoking, diabetes, and other risk factors to generate conventional lipid-rich atherosclerosis is markedly attenuated. These factors remain biologically important, however, because they can influence endothelial permeability, lipoprotein retention and modification, inflammation, thrombosis, and the clinical consequences of existing plaque.[1,2]
Mendelian randomization studies provide robust evidence for this relationship, demonstrating that lifelong, genetically predicted lower concentrations of ApoB-containing lipoproteins translate into a substantially greater reduction in coronary heart disease risk than the reductions achieved by short-term pharmacological trials initiated later in life.[1,23]
The “Athletic Atherosclerosis Hypothesis” proposes that chronic, lifelong endurance training alters the traditional risk-to-exposure relationship. Individuals who accumulate decades of high-volume aerobic exercise subject their coronary arteries to unusual mechanical conditions. During exercise, heart rate can remain above 150 to 180 beats per minute for hours, generating thousands of additional cardiac cycles per day, while cardiac output rises five- to six-fold. This high-flow, high-pressure state produces sustained elevations in epicardial blood-flow velocity and circumferential arterial wall deformation.
The primary question evaluated in this review is whether these mechanical forces accelerate the rate of transendothelial lipid infiltration and subendothelial retention, effectively lowering the threshold of cumulative ApoB exposure required to initiate and progress coronary plaque. If correct, maintaining lower circulating ApoB concentrations could provide an additive clinical benefit in protecting these highly fit individuals from subclinical vascular progression.
Scope and Approach
This manuscript is intentionally framed as a narrative review and critical appraisal rather than a systematic review: it does not document a reproducible search workflow (named databases, search strings, prespecified eligibility criteria, formal screening, or a PRISMA flow diagram) and should not be read as PRISMA-compliant. Where the argument moves from established biology to extrapolation, that transition is made explicit.
Three questions organize the appraisal. First, is there a biologically plausible pathway by which chronic endurance loading could facilitate ApoB entry or retention in the coronary wall? Second, do imaging data support a reproducible, athlete-specific phenotype of increased subclinical plaque or calcification? Third, if both propositions are at least partly true, what are the implications for lipid management in athletes whose baseline event risk may remain low despite detectable subclinical disease? Throughout, direct human coronary evidence is distinguished from cell-culture work, animal models, computational modeling, and inference.
Mechanical and Biomechanical Forces During Extreme Aerobic Training
The coronary vascular tree is continuously exposed to three-dimensional mechanical forces: blood-pressure-induced normal force (circumferential wall stress), blood-flow-induced tangential force (endothelial wall shear stress), and myocardial-contraction-induced cyclic deformation. During chronic endurance training these forces are amplified and sustained, generating a physical microenvironment distinct from that of sedentary individuals.
| ESTABLISHED DURING EXERCISE
Elevated coronary flow, heart rate, pulsatile pressure, and cyclic deformation ↓ ANATOMICALLY LOCALIZED POSSIBILITY Altered low / oscillatory / multidirectional shear or wall strain at geometrically susceptible bends and bifurcations ↓ (hypothesized; not demonstrated in athlete coronary arteries) HYPOTHESIZED ATHLETE-SPECIFIC RESPONSES Focal glycocalyx remodeling · altered endothelial transport · altered intimal extracellular matrix ↓ (hypothesized; not demonstrated in athlete coronary arteries) UNPROVEN CONSEQUENCE Greater entry or retention of ApoB-containing particles ↓ (hypothesized; not demonstrated in athlete coronary arteries) CLINICAL HYPOTHESIS Increased focal plaque initiation or progression in susceptible athletes |
At rest, normal flow patterns generate a physiological level of laminar shear stress that maintains endothelial health by stimulating nitric oxide (NO) production and downregulating adhesion molecules.[44] During extreme exertion, the epicardial arteries must accommodate high-flow, high-velocity perfusion. Computational fluid dynamics (CFD) modeling indicates that while time-averaged wall shear stress (TAWSS) remains elevated and globally protective across most of the coronary tree during high-output exercise, localized regions experience altered fluid dynamics.
Coronary bifurcations and curved segments exhibit spatially heterogeneous shear patterns because of their geometry. During exercise, increased flow raises wall shear stress over much of the coronary tree, which is generally considered atheroprotective. Whether exercise worsens low, oscillatory, or multidirectional shear at individual susceptible sites, improves it, or produces mixed effects depends on local anatomy, vessel motion, cardiac phase, and flow conditions, and has not been established longitudinally in endurance athletes. Where disturbed local hemodynamics do persist, they may impair endothelial homeostasis at the same focal “hot spots” that harbor plaque preferentially in the general population.[43]
Exercise-induced hypertension is a common but under-recognized phenomenon in masters and elite athletes. During maximal dynamic endurance exertion, systolic blood pressure can rise to roughly 200–220 mmHg in trained men and women, with values approaching 250 mmHg in selected highly trained athletes at the extreme upper end of effort (the upper extreme of reported observations rather than a typical response); heavy resistance exercise can transiently produce even higher pressures through a different, Valsalva-associated mechanism. These loads markedly increase transmural pressure and circumferential wall tension in epicardial vessels.[13] This high pulsatile pressure is a major determinant of vessel stretch and may contribute to repetitive deformation of endothelial cells and underlying vascular smooth muscle cells. Crucially, transient exercise hypertension is not equivalent to sustained chronic hypertension: the two differ fundamentally in duration (minutes to hours versus around the clock), pressure waveform, and the endothelial adaptations that accompany training. A peak of 200 mmHg during effort therefore carries very different biological implications than a resting pressure of 200 mmHg, and the hypothesis here concerns cumulative, intermittent loading rather than a chronic pressure overload.
Circumferential stretch triggers intracellular signaling pathways, activating mitogen-activated protein kinases (MAPK) and upregulating inflammatory mediators such as tumor necrosis factor-alpha (TNF-α). Repetitive, high-frequency physical stretch over decades of training imposes cumulative mechanical loading on the arterial wall, potentially altering compliance and predisposing the vessel to microvascular distortion, endothelial activation, and localized lipid accumulation.
At the molecular level, endothelial cells convert these mechanical inputs into biochemical signals through a defined mechanotransduction apparatus. The mechanosensitive cation channel Piezo1 is the best-characterized endothelial shear sensor, gating calcium influx in response to frictional force[40], while integrin–YAP/TAZ signaling translates flow direction into transcriptional programs. Importantly, unidirectional (laminar) shear engages an integrin–YAP/TAZ–JNK cascade that is atheroprotective, whereas disturbed, oscillatory flow shifts the same machinery—Piezo1, integrins, and YAP/TAZ—toward pro-inflammatory, atheroprone signaling[41]. This flow-pattern dependence reinforces the central point that the relevant risk is focal and disturbed-flow-specific rather than a global consequence of high flow. As with the other mechanistic pathways discussed here, these mechanotransduction cascades have not been directly demonstrated in the coronary arteries of healthy human endurance athletes and are invoked by analogy from vascular-biology models.
Mechanistic Review of the Response-to-Retention Model in Athletes
The response-to-retention hypothesis holds that the rate-limiting step in atherogenesis is the physical entrapment and retention of ApoB-containing lipoproteins within the subendothelial intimal space.[3,4,37] For a circulating lipoprotein to participate in plaque formation, it must traverse the vascular endothelium and interact with the extracellular matrix. This transendothelial passage and subsequent retention are highly regulated and are directly influenced by the mechanical stresses of chronic exercise.
Endothelial Glycocalyx Degradation (Shedding)
The first structural barrier to lipoprotein infiltration is the endothelial glycocalyx (eGC), a negatively charged, carbohydrate-rich, gel-like layer lining the luminal surface of endothelial cells.[5,39] Composed of proteoglycans (primarily syndecans and glypicans) linked to negatively charged glycosaminoglycan (GAG) chains (heparan sulfate and chondroitin sulfate), the eGC contributes to size-, charge-, and flow-dependent regulation of macromolecular access to the endothelial surface. Experimental and modeling studies suggest that an intact eGC can reduce LDL concentration and transport near the plasma membrane; it should not, however, be described as completely excluding LDL or preventing receptor-mediated interactions, since basal lipoprotein–endothelial interaction and transport occur in intact vessels.
A key mechanistic nuance must be stated precisely. Habitual exercise, taken as a whole, improves systemic endothelial function, raises nitric oxide bioavailability, and is broadly vasculoprotective; the hypothesis advanced here is emphatically not that endurance training harms the endothelium globally. Uniform, high laminar shear stress is itself generally glycocalyx-protective and stimulates its biosynthesis. Rather, it is the low, oscillatory, and disturbed shear found at bifurcations, arterial bends, and branch points—together with oxidative stress and elevated transmural pressure—that promotes eGC “shedding,” the enzymatic cleavage and release of syndecan-1, heparan sulfate, and hyaluronic acid into the circulation. The athletic hypothesis is therefore narrow and focal: not that high flow uniformly strips the glycocalyx, but that decades of repeated exposure to a limited number of disturbed-flow hot spots, superimposed on transient oxidative and pressure loads, could theoretically promote localized glycocalyx remodeling at the very segments already predisposed to plaque, while the remainder of the arterial tree benefits from exercise. It should be emphasized that the evidence for glycocalyx shedding is strongest for disturbed flow, oxidative injury, diabetes, hypertension, and inflammation, and that this focal remodeling has not been demonstrated in the coronary arteries of healthy human athletes.
If localized glycocalyx remodeling or thinning occurs at these sites, the transvascular permeability of the wall would be altered, allowing circulating ApoB-containing lipoproteins more direct access to the underlying endothelial cell membrane.
Transcellular Vesicular Transport (Transcytosis)
Because intact LDL particles are larger than conventional interendothelial junctional gaps (typically < 6 nm)—LDL particles are approximately 20–25 nm in diameter, with larger VLDL remnants extending up to ~80 nm—transcellular transport appears to be an important route across relatively intact arterial endothelium. This process, termed transcytosis, is mediated by caveolae (cholesterol-rich, flask-shaped plasma-membrane invaginations) and specific cargo receptors, with strong experimental support for caveolin-associated pathways involving scavenger receptor class B type 1 (SR-B1) and activin receptor-like kinase 1 (ALK1). Paracellular transport through transiently widened or leaky junctions can also contribute, particularly during endothelial turnover, inflammation, or injury, and the relative quantitative contributions of these pathways in human coronary arteries remain incompletely defined.[6,7,8]
In the cerebral vasculature, transcytosis is also mediated by the classical LDL receptor (LDLR). Basal transcytotic flux across systemic arterial endothelium occurs largely through LDLR-independent, caveolin-1–dependent pathways, although LDLR-dependent and inflammation-induced routes can contribute under specific conditions. The centrality of the caveolar route is underscored by genetic evidence: caveolin-1 knockout mice show markedly reduced LDL transport across the endothelium and are relatively protected from lesion formation despite hyperlipidemia. A paracellular route can also open transiently at sites of endothelial cell turnover, apoptosis, or mitosis (so-called leaky junctions), contributing to LDL entry alongside transcytosis.[9]
Mechanical strain and hydrostatic pressure can alter caveolar abundance, trafficking, and endothelial permeability in experimental systems. Caveolae participate in signaling, membrane buffering, and endocytosis, so a greater number of caveolae does not by itself establish greater vectorial transport of LDL; whether exercise-relevant coronary strain increases net luminal-to-intimal transport of ApoB-containing particles through SR-B1–, ALK1–, or caveolin-dependent pathways in vivo has not been established.
Extracellular Matrix Proteoglycan Synthesis and Affinity Modifications
Once a lipoprotein crosses the endothelial monolayer, its retention within the intima is determined by ionic binding to extracellular matrix proteoglycans synthesized by vascular smooth muscle cells (VSMCs). The small leucine-rich proteoglycan biglycan (BGN) is a predominant structural mediator of this retention, acting alongside other intimal proteoglycans such as versican, perlecan, and decorin. Biglycan carries negatively charged chondroitin sulfate and dermatan sulfate GAG chains that bind positively charged basic amino-acid residues on the surface of ApoB-100.[10]
Experimental mechanical strain can alter VSMC proteoglycan synthesis and GAG structure. In such models, sustained loading increases vascular smooth muscle cell (VSMC) synthesis and secretion of biglycan and decorin, driven principally by transforming growth factor-beta (TGF-β)/Smad mechanotransduction, with focal adhesion kinase (FAK)–associated signaling proposed as a contributing but less firmly established pathway. Whether the intermittent strain pattern of endurance exercise produces similar chronic changes in healthy human coronary intima is unknown.[11,12]
Mechanical strain can also modify GAG side-chain length, charge density, and sulfation in experimental systems, alterations that in vitro enhance the binding affinity of the extracellular matrix for ApoB-containing lipoproteins. The extent to which such changes occur in the coronary intima of healthy athletes, and their net effect on lipoprotein retention in vivo, remains undefined.
Established vs. Speculative Mechanobiological Pathways
In evaluating these adaptations, it is essential to separate established physiological observations from pathways that remain speculative in the specific context of the healthy human athlete.
| Established Vascular Mechanisms | Speculative Athletic Hypotheses |
| • The general response-to-retention pathway of atherogenesis.
• Glycocalyx shedding in response to oxidative/inflammatory stress and disturbed shear. • Caveolae-mediated active transcellular transport of LDL (SR-B1, ALK1). • In vitro / animal upregulation of biglycan under mechanical strain (TGF-β). |
• Direct in vivo documentation of exercise-induced local glycocalyx shedding in human coronary arteries.
• Exercise-specific proteoglycan GAG affinity modifications in healthy, elite human athletes. • Demonstration that mechanically driven retention outpaces metabolic clearance in non-atherosclerotic walls. |
Established mechanisms. The general response-to-retention model is widely accepted as the primary initiating event of ASCVD. The role of the glycocalyx as a selective permeability barrier, and its degradation under oxidative, inflammatory, and disturbed-flow conditions, is well documented, as is active transcellular LDL transport via caveolae and its dependence on SR-B1 and ALK1. Mechanical stimulation of VSMCs increasing proteoglycan synthesis and altering GAG sulfation has been robustly demonstrated in cell culture and animal stretch models.
Speculative hypotheses. Direct extrapolation of these cellular mechanisms to the coronary arteries of healthy, elite human endurance athletes remains speculative: in vivo visualization of coronary glycocalyx shedding and quantitative measurement of biglycan–ApoB binding affinity in healthy, active human athletes have not been performed, and whether these retention pathways would overcome the protective adaptations of exercise (detailed below) is unknown.
Competing Protective Mechanisms: Why Exercise May Also Reduce ApoB Entry
The retention-centered hypothesis must be weighed against the powerful, well-established vascular benefits of exercise, several of which act directly against lipoprotein entry and retention. Habitual endurance training raises endothelial nitric oxide bioavailability, improves flow-mediated dilation, and shifts endothelial gene expression toward an anti-inflammatory, anti-adhesive, atheroprotective phenotype.[45] Sustained high laminar shear stress—the dominant flow pattern across most of the coronary tree during exercise—stimulates glycocalyx biosynthesis rather than degradation, potentially reinforcing the very barrier whose focal loss the hypothesis invokes.
Beyond the endothelium, training lowers systemic and vascular inflammation (reduced hsCRP and pro-inflammatory cytokines), enhances antioxidant defenses and mitochondrial efficiency (limiting the oxidative stress that drives LDL modification and glycocalyx shedding, and lowering circulating oxidized LDL), and upregulates autophagy and endothelial repair programs that maintain barrier integrity. Exercise also improves HDL function and reverse cholesterol efflux capacity and lowers triglyceride-rich remnant lipoproteins—each reducing the pool and residence time of atherogenic particles available for subendothelial retention. Any one of these adaptations could, in principle, outweigh the proposed mechanical effect entirely.
These protective mechanisms are a principal reason endurance athletes enjoy markedly lower cardiovascular and all-cause mortality, and they plausibly offset much of any biomechanically driven increase in ApoB entry. The pertinent question is therefore not whether exercise protects the vasculature—it clearly does—but whether, at the extreme upper end of lifelong training volume, focal mechanical stresses at a limited number of disturbed-flow sites can locally outpace these globally protective adaptations.
Two observations suggest the balance is not always fully protective at the highest exposures. First, several—but not all—observational imaging studies of predominantly middle-aged and older male endurance athletes have reported higher coronary calcium or plaque prevalence than in carefully selected active controls, with heterogeneous findings regarding plaque composition and the relative roles of exercise volume and intensity; this suggests that the protective mechanisms, however potent, may not fully neutralize the plaque signal in such cohorts. Second, the protective effects are largely systemic and global, whereas the proposed retention risk is focal and anatomically concentrated at atheroprone segments; a globally healthier endothelium does not guarantee protection at every bifurcation and bend. This asymmetry between global protection and focal vulnerability is the crux of the hypothesis and the reason the two effects need not cancel. It nonetheless remains entirely possible that the protective mechanisms fully offset the mechanical risk, and that the observed plaque reflects the competing explanations discussed later rather than net harm; distinguishing these possibilities will require prospective mechanistic and outcome data. Stated plainly: despite these powerful protective mechanisms, observational imaging studies continue to demonstrate greater plaque burden in selected lifelong athletes, suggesting either incomplete compensation or an alternative explanation that remains unidentified.
Coronary Arterial Remodeling: Physiological Buffer vs. Subclinical Progression
Clinical evaluation of coronary disease in endurance athletes must account for the structural and functional adaptations of the athletic heart, commonly termed “exercise-induced coronary remodeling.” Epicardial arteries are dynamic structures that adapt to repetitive hemodynamic loads by altering caliber, wall thickness, and vasomotor responsiveness.
During training, sustained increases in coronary blood flow and laminar shear stress enhance endothelial-derived NO bioavailability, improving vasodilation and lowering resting vascular resistance. Over months to years this functional adaptation is supplemented by structural outward (positive) remodeling: quantitative coronary angiography, echocardiography, and cardiac MRI show that competitive endurance athletes possess significantly larger proximal coronary lumen diameters and cross-sectional areas than sedentary controls, proportional to the degree of eccentric left ventricular hypertrophy.
| The Protective Buffer | The Clinical Delay |
| • Sustained myocardial perfusion.
• Preserved coronary flow reserve. • High ischemic-symptom threshold. |
• Absence of classic stable angina.
• Subclinical plaque progresses unseen. • Possible sudden presentation of acute events. |
Endurance training improves coronary vasodilator capacity and myocardial perfusion efficiency and increases microvascular density. In individuals with established coronary disease, repeated ischemic stimuli and exercise training may enhance collateral function, but robust protective collateral networks capable of compensating for severe epicardial obstruction should not be presumed in otherwise healthy athletes. A larger resting lumen and improved vasodilator capacity nonetheless provide a physiological buffer that helps preserve coronary flow reserve, allowing athletes to perform at high workloads despite a meaningful burden of subclinical plaque.
This compensatory remodeling is, however, a double-edged sword. Glagov and colleagues showed that coronary segments enlarge to accommodate plaque until roughly 40% of the internal elastic lamina area is occupied before the lumen begins to narrow[42]. This compensation can preserve lumen dimensions during early plaque growth, allowing substantial disease to remain angiographically silent, so that the first clinical manifestation may be an abrupt acute event during extreme exertion. Two distinct processes should not be conflated: Glagov-type positive remodeling is a local response to plaque growth (and is itself a recognized feature of some high-risk plaques), whereas the generalized physiological enlargement of coronary arteries reported in trained athletes is an adaptation to chronic high flow. Positive remodeling is therefore not uniquely a protective athletic adaptation; what may distinguish the endurance athlete is the combination of expanded coronary caliber and a high ischemic threshold, which together can delay clinical recognition.
Review of Coronary Imaging Studies in Endurance Athletes
Modern sports cardiology relies on multi-modality coronary imaging—CAC scoring, coronary CT angiography (CCTA), intravascular ultrasound (IVUS), and optical coherence tomography (OCT)—to characterize coronary disease in highly trained individuals.
Coronary Artery Calcium (CAC) Scoring and Plaque Distribution
Early evaluations used non-contrast CT to quantify CAC with the Agatston method. Several observational studies have reported higher CAC among highly active individuals and masters athletes relative to age-matched controls. In a 25-year follow-up of 3,175 CARDIA participants, individuals exceeding physical activity guidelines had a higher likelihood of a CAC score above zero (adjusted OR 1.86, 95% CI 1.16–2.98), an association most pronounced among white male participants.[18]
Similarly, Sung and colleagues studied 25,485 healthy, asymptomatic adults and found that those classified as “health-enhancing physically active” (HEPA) exhibited higher baseline CAC and more rapid progression of calcification over follow-up than inactive individuals.[19]
Among dedicated athletic cohorts, the Marathon Study (Möhlenkamp et al.) evaluated 108 male marathon runners (aged ≥ 50 years, ≥ 5 marathons in the prior 3 years) against 864 controls drawn from the Heinz Nixdorf Recall population cohort, matched 8:1 by age and 2:1 by Framingham risk. Detectable CAC was present in 71% of runners, and 36% had a CAC score > 100—significantly higher than in risk-matched controls.[17]
In a UK cohort of 152 masters athletes (77% runners, 23% cyclists) versus 92 controls (mean age 54 years), Merghani and colleagues found severe coronary disease (CAC ≥ 300) in 11.3% of male athletes versus none of the risk-matched controls (P = 0.009); male athletes exhibited predominantly calcified plaques, whereas sedentary controls showed predominantly mixed plaques.[16]
High-Resolution Plaque Tissue Characterization (CCTA, IVUS, OCT)
Advanced CCTA and intravascular imaging permit high-resolution characterization of non-calcified, mixed, and high-risk plaque, revealing that the coronary plaque profile of athletes is heterogeneous.
| Densely Calcified Plaques | Vulnerable Soft Plaques |
| • High attenuation (> 130 HU).
• Compositionally stable, thick fibrous cap. • Low risk of mechanical rupture. • Relatively promoted by very-vigorous training. |
• Low attenuation (< 30 HU), large necrotic core.
• Non-calcified proximal lesions; napkin-ring sign; TCFA. • Higher absolute burden in lifelong athletes than in active controls. • Progress silently under positive remodeling. |
Calcified plaques. Calcified plaque is conventionally identified above approximately 130 HU on non-contrast CAC imaging, but 130 HU is the minimum threshold used to detect calcium and should not be equated with densely calcified plaque; higher calcium density may reflect more mature or healed plaque, although its prognostic meaning depends on total calcium volume, plaque burden, and clinical context. Greater macrocalcification is often interpreted as a marker of more mature or healed disease, but CAC alone does not establish fibrous-cap thickness, exclude coexisting non-calcified components, or render a lesion clinically benign.
Mixed and non-calcified plaques. The assumption that athletes develop only “safe,” heavily calcified plaques is incorrect. Using high-resolution CCTA, the Master@Heart study showed that lifelong masters endurance athletes harbor a significantly higher absolute burden of mixed and non-calcified plaque than active, non-athletic controls—any coronary plaque was present in 63.4% of lifelong athletes versus 50.0% of controls, and any proximal plaque in 55.5% versus 40.3%. Lifelong athletes had higher odds of at least one non-calcified plaque (23.6% vs. 15.3% of controls; OR 1.95, 95% CI 1.12–3.40) and at least one non-calcified proximal plaque (16.8% vs. 7.4%; OR 2.80, 95% CI 1.39–5.65).[14]
Fibrous plaques. Between the densely calcified and lipid-rich non-calcified extremes lies the fibrous plaque—a lesion rich in smooth muscle cells and collagen-dense extracellular matrix, with intermediate CT attenuation and comparatively little lipid or calcium. Fibrous and fibrocalcific lesions are generally regarded as biologically quiescent and mechanically stable, and they may represent a healed or stabilizing phenotype.[38] Their prominence in some athletic cohorts is consistent with the interpretation that much of the athlete plaque burden reflects a shift toward more stable morphologies over time, even as the absolute lesion count rises.
Vulnerable-plaque features. Vulnerable plaques are marked by low-attenuation plaque (< 30 HU), positive remodeling, microcalcification, and the napkin-ring sign. Because the major athlete-imaging studies used CAC scoring and CCTA rather than routine intracoronary NIRS or OCT, direct comparisons of thin-cap fibroatheroma prevalence in healthy endurance athletes are limited. What can be said is that the elevated burden of non-calcified and mixed proximal lesions reported in some athletic cohorts indicates that athletic status does not confer absolute protection against potentially unstable plaque.
Clinical Outcome Studies, Longevity, and Cardioprotective Redundancies
To reconcile an increased subclinical plaque burden with superior fitness, long-term outcome registries are informative. Highly active individuals, masters athletes, and elite competitors consistently show reductions in both cardiovascular and all-cause mortality relative to the general population. In a cohort of more than 21,000 men stratified by physical activity and CAC, the most active individuals had lower all-cause and cardiovascular mortality across all CAC categories; highly active men with CAC ≥ 100 had a mortality hazard at or below that of inactive men with a CAC score of zero—an observation suggesting that high fitness may attenuate the prognostic weight of calcification, but which should not be read to mean that calcified plaque is benign.[20] This survival advantage is especially pronounced in elite cohorts: French Tour de France participants have shown roughly 41% lower all-cause and 33% lower cardiovascular mortality than the general male population, with correspondingly greater average longevity.[21,22]
The cardioprotection of endurance athletes reflects several integrated, redundant adaptations:
- Autonomic regulation. Training-related autonomic adaptation lowers resting heart rate and improves heart-rate recovery, both generally associated with favorable prognosis. Its effect on arrhythmic risk is not uniformly protective, however: lifelong endurance training may also increase susceptibility to atrial arrhythmias (notably atrial fibrillation) and, in selected individuals, to clinically important brady- or ventricular arrhythmias.
- Myocardial efficiency and compliance. Physiological eccentric left ventricular hypertrophy increases end-diastolic volume and stroke volume and permits a lower heart rate at a given submaximal external workload, improving whole-body exercise efficiency. This should not be interpreted as reducing myocardial oxygen demand during maximal exertion, when demand is very high.
- Metabolic and anti-inflammatory homeostasis. High insulin sensitivity, efficient mitochondrial substrate oxidation, favorable blood pressure, and low circulating hsCRP and pro-inflammatory cytokines minimize the probability of plaque activation and destabilization.
- Vascular compliance and perfusion redundancy. High NO bioavailability, anti-atherogenic gene expression, and preserved arterial compliance support myocardial perfusion; in selected individuals with coronary disease, exercise training may also improve collateral function, though robust collateral networks should not be presumed in otherwise healthy athletes.
These redundancies substantially reduce the clinical consequences of subclinical plaque, but the safety margin is finite. In susceptible individuals with underlying coronary disease, vigorous exertion can transiently increase the risk of an acute event through rises in myocardial demand, catecholamines, blood pressure, and platelet activity, and possibly plaque-related mechanisms; the absolute risk remains low in regularly trained individuals, and direct mechanical plaque disruption by exercise should not be presumed.
Genetic Architecture of ApoB and Cumulative Lifetime Particle Exposure
Modern lipid management increasingly focuses on the absolute particle concentration of ApoB-containing lipoproteins rather than cholesterol cargo such as LDL-C. Each hepatically derived atherogenic particle—VLDL, IDL, LDL, and Lp(a)—contains one ApoB-100 molecule, whereas intestinal chylomicron and remnant particles contain one ApoB-48 molecule; clinical ApoB assays capture both forms, although fasting ApoB is dominated by ApoB-100-containing particles. Measuring ApoB therefore approximates the total number of atherogenic particles capable of crossing the endothelium and initiating atherogenesis.[2]
When LDL-C and ApoB are discordant—as in insulin resistance, obesity, or elevated triglycerides, where small, dense LDL particles predominate—risk often tracks more closely with ApoB (or non-HDL-C) than with LDL-C, so ApoB can provide important incremental information, particularly in hypertriglyceridemia, diabetes, obesity, and metabolic syndrome.
| The causal mechanism of atherogenesis
↓ Total particle number (ApoB) > cholesterol-mass cargo (LDL-C) ↓ Strong causal evidence (MR) | Cumulative-exposure model genetically lower ApoB → lifelong, risk ≈ magnitude × duration; unbiased protection (LDLR, PCSK9, early control prevents APOC3) subendothelial retention |
Mendelian-randomization analyses provide strong causal evidence for this particle-centric model by using inherited genetic variants as instruments for lifelong differences in lipoprotein exposure. These analyses remain dependent on instrument validity and assumptions regarding pleiotropy and population structure, and so should not be described as literal proof or as completely unbiased experiments. Genetic scores mimicking HMG-CoA reductase, PCSK9, NPC1L1, and APOC3 inhibition show a consistent, log-linear relationship between cumulative ApoB exposure and coronary heart disease risk. Variant analyses of triglyceride-lowering (APOC3/LPL) and LDL-lowering (LDLR/PCSK9) pathways indicate that, once scaled to the same ApoB reduction, each confers a comparable magnitude of coronary risk reduction, and that combined exposure is additive.[23] Randomized-trial evidence is concordant: the Cholesterol Treatment Trialists’ Collaboration meta-analyses show an approximately 20–25% proportional reduction in major vascular events per 1 mmol/L (≈38.6 mg/dL) reduction in LDL-C over typical follow-up; the corresponding ApoB change varies by baseline phenotype and treatment and is not represented by a fixed conversion.[24]
For the athletic population, this genetic architecture carries a clear implication: athletes possess exceptional fitness and metabolic health but do not inherit immunity to lipid abnormalities. Dyslipidemia is common and frequently under-recognized in competitive athletes, with screening of Olympic and Paralympic competitors identifying elevated LDL-C or other lipid abnormalities (depending on the threshold used) in roughly 32% to 36%.
Because mutations reducing LDL-receptor function or increasing ApoB production generate a high particle burden from birth, cumulative lifetime exposure in these athletes is substantially elevated independent of training volume.[32,33] In the absence of pharmacological intervention, a genetically high particle count—combined with any mechanically enhanced transcytosis and intimal retention during extreme exercise—can accelerate subclinical plaque progression and, in principle, erode the exercise-induced safety buffer.
Pharmacological Interventions and Therapeutic Management in Athletes
When an endurance athlete presents with established coronary disease, elevated CAC, or high ApoB that cannot be managed by lifestyle alone, pharmacological therapy should be considered within current guideline recommendations. Managing lipids in competitive and masters athletes requires careful attention to drug side effects, training adaptations, and exercise tolerance.
Statins and Statin-Associated Muscle Symptoms (SAMS)
Statins are the primary agents for lowering ApoB and reducing cardiovascular events. By competitively inhibiting hepatic HMG-CoA reductase, they upregulate cell-surface LDL receptors and enhance clearance of ApoB-containing particles.
Their utility in athletes is frequently challenged by statin-associated muscle symptoms (SAMS). It is important to distinguish objectively defined statin myopathy—marked creatine kinase elevation (> 10× the upper limit of normal), and, more rarely still, rhabdomyolysis—which is genuinely uncommon, affecting well under 0.1% of users, from the far more prevalent syndrome of subjective muscle aches, weakness, and cramps without major CK elevation. Blinded randomized evidence finds only a small excess of predominantly mild symptoms attributable to statins, concentrated early after initiation, whereas unblinded observational studies and registries report much higher symptom frequencies, sometimes approaching 10%–30%—estimates strongly affected by definitions, selection, and expectation (nocebo) effects. Whether competitive athletes have a substantially higher pharmacologically caused SAMS rate remains uncertain, though they may report symptoms more often given high training loads, muscle microtrauma, and heightened somatic awareness.
Proposed mechanisms include altered mitochondrial energetics and reduced isoprenoid (including coenzyme Q10) synthesis, oxidative stress, and disturbed intracellular calcium handling; their relative importance in typical statin-associated symptoms remains uncertain, and CoQ10 depletion has not been shown to establish that supplementation prevents symptoms. Under the repetitive eccentric loads of endurance exercise, such factors could in principle exacerbate soreness or prolong recovery in susceptible individuals, but this is not established.
High-dose lipophilic statin therapy (for example, atorvastatin 40–80 mg or simvastatin 40 mg) may blunt mitochondrial biogenesis and diminish expected gains in VO₂peak and muscle citrate-synthase activity during aerobic training. In the STOMP trial, atorvastatin 80 mg/day for 6 months in healthy, statin-naive subjects produced a modest mean creatine kinase increase (+20.8 U/L; P < 0.0001) and slightly more myalgia than placebo, without objectively impairing muscle strength or exercise capacity.[31] To minimize SAMS while achieving lipid goals, a structured, stepwise strategy is advised.
| Masters athlete with indication for ApoB lowering
↓ Step 1: low-dose hydrophilic statin + ezetimibe rosuvastatin 5–10 mg or pravastatin; add ezetimibe 10 mg ↓ Well tolerated → achieve target | SAMS → assess timing/causes, CK if indicated, brief interruption then rechallenge ↓ Step 2: non-statin phase bempedoic acid 180 mg · add/escalate to PCSK9 inhibitor (mAb or inclisiran) |
Statin selection and dosing. For an athlete experiencing muscle symptoms, reasonable options include a lower dose, alternate-day administration, or a different statin. Hydrophilic agents such as pravastatin or rosuvastatin are sometimes selected empirically because of lower passive skeletal-muscle penetration, but a clinically meaningful muscle-sparing advantage over lipophilic statins at equivalent lipid-lowering potency has not been conclusively demonstrated.
Combination therapy with ezetimibe. To avoid high-dose statin monotherapy, early combination with low-dose ezetimibe (10 mg)—which blocks the NPC1L1 transporter in the small intestine—adds an incremental LDL-C reduction of roughly 23% to 24% when combined with a statin (with a somewhat smaller reduction in ApoB on a percentage basis). A low-dose statin plus ezetimibe can produce substantial additional LDL-C and ApoB lowering while avoiding escalation to the highest statin doses; the magnitude varies by regimen and baseline phenotype, and preservation of athletic performance has not been established in dedicated trials.[27]
Oral and Injectable Non-Statin Alternatives
For athletes with documented statin intolerance, or who fail to reach ApoB targets on maximally tolerated therapy, several effective non-statin options exist.
Bempedoic acid. Bempedoic acid is a first-in-class oral prodrug inhibiting adenosine triphosphate–citrate lyase (ACLY), upstream of HMG-CoA reductase. It requires enzymatic activation by very-long-chain acyl-CoA synthetase-1 (ACSVL1), which is highly expressed in liver but essentially absent in skeletal muscle; consequently it cannot be activated within myofibers. It provides robust lipid lowering (LDL-C ~21% and ApoB ~15% as monotherapy) and anti-inflammatory effects (hsCRP reduced by 22.2%) with a rate of myalgia statistically indistinguishable from placebo. It can, however, raise uric acid and the risk of gout and has been associated with small increases in cholelithiasis and tendon-related adverse events; tendon symptoms are particularly relevant when counseling competitive athletes, even though the absolute risk is low. Its outcome benefit was demonstrated in statin-intolerant high-risk patients (CLEAR Outcomes), not specifically in healthy endurance athletes.[25,26]
In the CLEAR Outcomes trial (13,970 statin-intolerant patients), bempedoic acid significantly reduced the primary 4-component MACE endpoint (HR 0.87, 95% CI 0.79–0.96; P = 0.004) and coronary revascularization (by 19%), confirming its therapeutic efficacy.[25]
PCSK9-directed therapies. Alirocumab and evolocumab are monoclonal antibodies that prevent lysosomal degradation of hepatic LDL receptors, commonly reducing LDL-C by ~50–60% or more, with somewhat smaller ApoB reductions, and have demonstrated cardiovascular outcome reductions in secondary-prevention trials, with muscle-related side effects at very low rates.[28,29] Inclisiran, a small interfering RNA that inhibits hepatic PCSK9 synthesis, is dosed on day 1, day 90, and every 6 months thereafter, producing sustained LDL-C and ApoB lowering with twice-yearly maintenance dosing; its definitive cardiovascular outcome evidence should be distinguished from the completed outcomes evidence for the PCSK9 monoclonal antibodies.[30]
Clinical Guidelines and Consensus Statements
Despite the expanding literature on subclinical plaque and accelerated calcification in endurance athletes, current major guidelines from the American Heart Association (AHA), American College of Cardiology (ACC), European Society of Cardiology (ESC), and European Atherosclerosis Society (EAS) do not define athlete-specific lipid or ApoB targets. Both athletes and sedentary individuals are stratified into risk categories using standardized equations such as the PREVENT-ASCVD equations or SCORE2.[34,36]
Guideline frameworks differ by jurisdiction. European (ESC/EAS) guidelines use explicit LDL-C and ApoB goals by risk category, including an LDL-C goal below 55 mg/dL for many very-high-risk patients; U.S. guidelines have historically used treatment thresholds and percentage lowering and, in the 2026 framework, restored treatment goals in specified categories. Importantly, the classification of asymptomatic CCTA plaque depends on plaque burden, stenosis, and clinical context, and non-obstructive plaque should not automatically be labeled equivalent to established clinical ASCVD. Across frameworks, ApoB is used as an adjunct to guide intensification once LDL-C and non-HDL-C goals are met, and a high CAC score (Agatston > 100 or > 75th percentile for age and sex) can prompt or intensify preventive therapy.[34]
Sports-cardiology groups have begun to address the physiology of lifelong runners and cyclists. A 2026 Swiss Olympic/Swiss Society of Sports Medicine (SEMS) practical framework—not a multinational, evidence-graded guideline—proposes an athlete-focused approach to lipid screening and treatment tolerance, highlighting several considerations:
- Pragmatic primary prevention. Athletes with clear, guideline-based indications for lipid lowering should not be denied evidence-based therapy purely because of theoretical concerns about performance or muscle soreness.
- Stepwise SAMS avoidance. To preserve training quality, prioritize low-dose hydrophilic statins (rosuvastatin), early ezetimibe combination, or muscle-sparing alternatives (bempedoic acid, PCSK9 inhibitors) in symptomatic individuals.
- Routine lipoprotein(a) screening. Consistent with the 2026 ACC/AHA multisociety dyslipidemia guideline, a one-time measurement of Lp(a) is recommended in all competitive and masters athletes. Because elevated Lp(a) (> 50 mg/dL or > 125 nmol/L) is an independent, genetically determined risk factor unaffected by exercise, its identification is crucial to personalize risk estimation.[34,35]
Methodological Appraisal of Major Observational Imaging Studies
To gauge the strength of the evidence bearing on the athletic-atherosclerosis hypothesis, the most influential imaging studies in this field—all observational rather than randomized—are appraised below using a structured narrative certainty assessment. Study design and population characteristics are summarized in Table 1; principal findings, methodological limitations, and certainty ratings appear in Table 2. (The landscape tables follow on the next pages.)
Table 1. Design and population characteristics of major coronary-imaging studies in endurance athletes
| Study / Trial | Design, population & N | Training & exercise metrics | Adjustments & statistical model |
| MARC-2 (2023) [15] | • Prospective cohort
• 289 male amateur athletes (median age 54; IQR 50–60) • Follow-up 6.3 ± 0.5 y |
• Median 41 MET-h/wk
• Moderate 0%; vigorous 44%; very vigorous 34% |
• Linear / logistic regression
• Adjusted: age, SBP, DBP, BMI, lipids, HbA1c, family history, baseline CAC |
| Master@Heart (2023) [14] | • Prospectively recruited, cross-sectional CCTA comparison
• 191 lifelong + 191 late-onset masters athletes; 176 active controls • All male, median age 55 |
• Lifelong 11 h/wk; late-onset 10 h/wk; controls 1 h/wk
• Fitness quantified via VO₂peak |
• Adjusted: age, BMI, blood pressure, lipids, smoking, family history, cardiorespiratory fitness |
| Merghani et al. (2017) [16]
(UK Masters cohort) |
• Cross-sectional comparative
• 152 masters athletes (77% runners, 23% cyclists) vs 92 controls • Mean age 54; low baseline risk |
• High-intensity training over decades
• Risk-factor-matched controls |
• Adjusted for baseline Framingham risk score |
| Marathon Study / Möhlenkamp et al. (2008) [17] | • Observational cohort
• 108 male marathon runners (≥ 50 y) vs 864 Heinz Nixdorf Recall controls |
• ≥ 5 marathons in prior 3 y
• Decades of recreational training |
• Matched 8:1 by age, 2:1 by Framingham risk
• Adjusted for individual risk factors |
Table 2. Principal findings, methodological limitations, and narrative certainty
| Study | Primary findings (HR / OR) | Limitations & bias risk | Funding / COI & narrative certainty |
| MARC-2 (2023) | • Total exercise volume not associated with plaque progression
• Vigorous: β = −0.05 per 10% increase (P = 0.02) • Very vigorous: β = +0.05 per 10% increase (P = 0.01) • Calcified plaque aOR 2.09 (P ≈ 0.03), highest vs lowest tertile |
• Exclusively male and White
• Self-reported exercise questionnaire • No clinical hard endpoints |
Academic / institutional grants; no commercial COI.
Certainty: low-to-moderate for association with plaque progression; no evidence on causality or athlete event risk. |
| Master@Heart (2023) | Lifelong athletes vs controls:
• ≥ 1 plaque OR 1.86 (1.17–2.94) • ≥ 1 proximal plaque OR 1.96 (1.24–3.11) • ≥ 1 non-calcified plaque OR 1.95 (1.12–3.40) • ≥ 1 non-calcified proximal plaque OR 2.80 (1.39–5.65) |
• Cyclist-dominant (77% cyclists)
• White males only • No longitudinal event tracking |
Public research grants (KU Leuven); no COI.
Certainty: low-to-moderate for the association with greater plaque prevalence in this selected population; no evidence on clinical outcomes or causality. |
| Merghani et al. (2017) | • Higher CAC in athletes
• CAC ≥ 300 in 11.3% of male athletes vs 0% of controls (P = 0.009) • Athletes predominantly calcified plaque; controls predominantly mixed |
• Small sample size
• Selection bias (recruited from athletic clubs) • No clinical outcomes |
British Heart Foundation; no industry COI.
Certainty: low for association (small, selected sample); no outcome data. |
| Marathon Study / Möhlenkamp (2008) | • Detectable CAC 71% (runners) vs lower in controls
• CAC > 100 in 36% of runners • CAC graded risk of all-cause mortality across categories |
• Self-selected athletic cohort
• Historical (2008) imaging technology • Male-only; no plaque-tissue characterization |
Public and foundation funding; no commercial COI.
Certainty: low-to-moderate for association (older imaging, male-only); limited outcome linkage. |
Note: The previously separate “UK Masters Study (2017)” and “Merghani et al. (2017)” entries describe the same cohort (152 athletes; 77% runners, 23% cyclists; vs 92 controls) and have been consolidated into a single reference [16]. The 108 marathon runners are attributed to the Marathon Study (Möhlenkamp et al., 2008 [17]); the Heinz Nixdorf Recall study supplied the matched control population.
Narrative Synthesis of Study Quality and Bias
A critical review of the major imaging studies reveals several recurring limitations:
- Selection and survivor bias. All major athletic imaging studies are susceptible to healthy-user selection bias: those who undertake extreme endurance exercise are inherently healthier, with lower rates of metabolic syndrome and superior fitness. Because these studies enroll older masters athletes (typically ≥ 50–55 years), they are also subject to survivor bias—athletes prone to early unstable-plaque rupture or malignant arrhythmia would already have been removed from the cohort, leaving survivors with predominantly stable, calcified coronary trees.
- Homogeneity of study populations. The cohorts are exceptionally homogeneous. In both MARC-2 and Master@Heart, participants were exclusively White males, limiting generalizability. Female athletes show different plaque characteristics—lower calcification, lower total plaque volume, and lower baseline risk. The cyclist-dominant Master@Heart cohort (77% cyclists) may also experience different mechanical coronary stresses than running-dominant cohorts.
- Lack of longitudinal outcome data. MARC-2 and Master@Heart provide high-resolution cross-sectional imaging but no longitudinal hard-outcome data. It remains a critical gap to determine whether the non-calcified and mixed proximal plaques seen in lifelong athletes carry the same hazard for myocardial infarction and sudden death as in sedentary, risk-matched cohorts.
Competing Explanations for the Athlete Plaque Paradox
Before attributing the athlete plaque signal to any specific mechanobiological pathway, it is essential to weigh alternative interpretations, several of which could partly or wholly account for the observed associations:
- Detection and surveillance bias. Health-conscious athletes undergo cardiac imaging more often than the general population, so a higher measured plaque prevalence may partly reflect ascertainment rather than a true excess.
- Calcification as stabilization. A higher calcified-plaque burden may represent healed, stabilized lesions—an endpoint of plaque repair—rather than more active or dangerous disease; CAC then marks past healing, not accelerated atherogenesis.
- Detectability (measurement hypothesis, unvalidated). It has been proposed that outward remodeling and the larger caliber of athletic arteries could affect plaque conspicuity on CCTA; larger vessels may reduce partial-volume effects and improve image quality, but the claim that they systematically inflate apparent plaque burden is not established and should be regarded as a measurement hypothesis requiring validation.
- Genetic predisposition. Endurance sport self-selects individuals with particular physiologies; unmeasured genetic factors (including Lp(a) and familial lipid traits) could confound the exercise–plaque association.
- Dietary and behavioral factors. High-volume training is often accompanied by very high caloric intake and specific dietary patterns whose long-term vascular effects are not captured by standard risk factors.
- Historical risk-factor exposure. In older masters cohorts, prior smoking and earlier-life risk-factor exposure predating athletic careers may contribute to present-day plaque independent of training.
- Selection and survivor bias. As discussed above, healthy-user selection and the survivorship of older athletes both shape these cross-sectional cohorts.
None of these alternatives is mutually exclusive with the mechanobiological hypothesis, and the truth likely involves several acting together. Acknowledging them explicitly is essential to avoid over-attributing the signal to a single, unproven pathway.
Limitations
Three limitations define the proper status of this review. First, direct mechanistic confirmation in living human athlete coronary arteries is lacking: the hypothesized roles of glycocalyx disruption, upregulated transcytosis, and proteoglycan remodeling in endurance athletes remain plausible extrapolations from cell, animal, and modeling work rather than demonstrated coronary findings in vivo. Second, the athlete-imaging literature is constrained by selection bias, survivor bias, modest sample sizes in several cohorts, and population homogeneity—much of it drawn from older White male masters athletes—so the proposed phenotype should not be generalized to women, younger athletes, or more diverse groups without caution. Third, athlete-specific outcome trials are absent: there is at present no randomized evidence that a lower ApoB threshold improves hard clinical outcomes specifically in asymptomatic endurance athletes with subclinical plaque. These limitations do not negate the thesis; they fix its status as hypothesis-generating and clinically pragmatic rather than prescriptive.
Conclusion
The current literature supports a plausible, hypothesis-generating, non-binary model: decades of high-volume aerobic exercise do not confer complete immunity from coronary disease, and the distinctive hemodynamic forces of extreme training may, at anatomically susceptible sites, contribute to localized changes in the vascular wall.
This microenvironment could plausibly increase the probability that circulating ApoB-containing lipoproteins traverse the endothelium and are retained within the epicardial wall, potentially facilitating subclinical plaque initiation and progression. Athletes are nonetheless substantially protected from clinical events by high cardiorespiratory fitness, favorable systemic risk factors, improved endothelial function, and myocardial reserve—adaptations that can act as a buffer, delaying symptoms even where subclinical plaque is present.
On mechanistic and genetic grounds, lowering circulating ApoB reduces the primary substrate for subendothelial retention. Whether lower ApoB targets provide incremental benefit beyond current guideline-directed lipid lowering in endurance athletes nonetheless remains unknown and should be evaluated in randomized clinical trials; because long-term outcome trials in asymptomatic athletes are lacking, any athlete-specific target remains hypothesis-generating. Athletes with documented plaque, elevated CAC, familial hypercholesterolemia, elevated Lp(a), diabetes, or other established risk enhancers should receive guideline-based risk assessment and lipid management according to the same principles applied to non-athletes, rather than reassurance based solely on high fitness. Treatment should follow contemporary prevention guidelines and shared decision-making; in athletes with muscle symptoms or performance concerns, reasonable individualized options include dose adjustment, a different statin, combination therapy with ezetimibe, or evidence-based non-statin therapy chosen according to baseline risk, plaque burden, expected absolute benefit, adverse-effect profile, access, and patient preference. It must be stated plainly that no current ACC, AHA, ESC, or EAS guidance endorses athlete-specific ApoB or LDL-C targets, and whether athletic exposure itself justifies lower targets at an otherwise identical level of plaque and clinical risk is unknown.
Diagnostic and Prevention Gaps, Unanswered Questions, and Future Directions
- Determining the true event rate of subclinical proximal plaque in masters athletes. Large, multicenter, prospective longitudinal registries are needed to track myocardial infarction, acute coronary syndrome, and sudden death in asymptomatic athletes with documented proximal plaque, and to determine whether high fitness and anti-inflammatory preconditioning stabilize these lesions over time.
- Establishing non-invasive biomarkers of coronary glycocalyx integrity. Validation of circulating biomarkers (syndecan-1, heparan sulfate, hyaluronic acid) or advanced microvascular imaging (e.g., sublingual dark-field microscopy) would allow tracking of glycocalyx degradation and recovery across exercise intensities and training volumes.
- Conducting dedicated randomized trials of muscle-sparing pharmacotherapy. Trials designed specifically for competitive and masters athletes should compare the efficacy, safety, and training impact of non-statin regimens (bempedoic acid, PCSK9 monoclonal antibodies, inclisiran) against standard high-dose statin monotherapy, with VO₂peak and recovery kinetics as endpoints.
- Testing athlete-specific ApoB targets in randomized trials. Randomized trials comparing ApoB or LDL-C targets (for example, LDL-C < 50 vs. < 70 mg/dL) in endurance athletes with documented coronary plaque are needed to determine whether more intensive lowering improves outcomes without compromising training tolerance—the central clinical question raised by this review.
- Characterizing subclinical CAD in diverse and female athletic cohorts. Prospective CCTA and intravascular-imaging studies in large female and ethnically diverse cohorts are needed to map vascular remodeling, plaque morphology, and event rates, enabling athlete-specific risk models (such as a proposed “Lipid Athlete Score”) that replace the generalizations of standard primary-prevention calculators.
Disclosures
Author contributions. P.M. is the sole author and is responsible for the conception, literature appraisal, drafting, and final approval of the manuscript, and accepts full responsibility for its content.
Funding. This work received no specific grant from any public, commercial, or not-for-profit funding agency.
Competing interests. The author is the founder and operator of Curing Heart Disease, LLC, a cardiovascular-health education platform (curingheartdisease.com) that publishes content on the topics discussed in this review. The author has no other financial or non-financial competing interests to declare.
Data availability. No new data were generated or analyzed in this narrative review; all data discussed derive from the cited published sources.
Ethics. This article is a review of previously published literature and did not involve new studies of human or animal subjects; institutional review board approval was therefore not required.
Use of AI. AI-assisted tools were used for language editing, organization, and source verification. All substantive claims, citations, interpretations, and final wording were reviewed and approved by the author, who accepts full responsibility for the manuscript. No AI tool is listed as an author. This disclosure is provided in accordance with current ICMJE recommendations; the author should adapt it to the target journal’s specific policy.
References
- Ference BA, Ginsberg HN, Graham I, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2017;38(32):2459-2472. doi:10.1093/eurheartj/ehx144
- Sniderman AD, Thanassoulis G, Glavinovic T, et al. Apolipoprotein B Particles and Cardiovascular Disease: A Narrative Review. JAMA Cardiol. 2019;4(12):1287-1295. doi:10.1001/jamacardio.2019.3780
- Williams KJ, Tabas I. The response-to-retention hypothesis of early atherogenesis. Arterioscler Thromb Vasc Biol. 1995;15(5):551-561. doi:10.1161/01.atv.15.5.551
- Tabas I, Williams KJ, Borén J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 2007;116(16):1832-1844. doi:10.1161/CIRCULATIONAHA.106.676890
- Reitsma S, Slaaf DW, Vink H, van Zandvoort MA, oude Egbrink MG. The endothelial glycocalyx: composition, functions, and visualization. Pflugers Arch. 2007;454(3):345-359. doi:10.1007/s00424-007-0212-8
- Huang L, Chambliss KL, Gao X, et al. SR-B1 drives endothelial cell LDL transcytosis via DOCK4 to promote atherosclerosis. Nature. 2019;569(7757):565-569. doi:10.1038/s41586-019-1140-4
- Kraehling JR, Chidlow JH, Rajagopal C, et al. Genome-wide RNAi screen reveals ALK1 mediates LDL uptake and transcytosis in endothelial cells. Nat Commun. 2016;7:13516. Published 2016 Nov 21. doi:10.1038/ncomms13516
- Zhang X, Sessa WC, Fernández-Hernando C. Endothelial Transcytosis of Lipoproteins in Atherosclerosis. Front Cardiovasc Med. 2018;5:130. Published 2018 Sep 25. doi:10.3389/fcvm.2018.00130
- Cancel LM, Tarbell JM. The role of apoptosis in LDL transport through cultured endothelial cell monolayers. Atherosclerosis. 2010;208(2):335-341. doi:10.1016/j.atherosclerosis.2009.07.051
- O’Brien KD, Olin KL, Alpers CE, et al. Comparison of apolipoprotein and proteoglycan deposits in human coronary atherosclerotic plaques: colocalization of biglycan with apolipoproteins. Circulation. 1998;98(6):519-527. doi:10.1161/01.cir.98.6.519
- Lee RT, Yamamoto C, Feng Y, et al. Mechanical strain induces specific changes in the synthesis and organization of proteoglycans by vascular smooth muscle cells. J Biol Chem. 2001;276(17):13847-13851. doi:10.1074/jbc.M010556200
- O’Callaghan CJ, Williams B. Mechanical strain-induced extracellular matrix production by human vascular smooth muscle cells: role of TGF-beta(1). Hypertension. 2000;36(3):319-324. doi:10.1161/01.hyp.36.3.319
- Caselli S, Vaquer Segui A, Quattrini F, et al. Upper normal values of blood pressure response to exercise in Olympic athletes. Am Heart J. 2016;177:120-128. doi:10.1016/j.ahj.2016.04.020
- De Bosscher R, Dausin C, Claus P, et al. Lifelong endurance exercise and its relation with coronary atherosclerosis. Eur Heart J. 2023;44(26):2388-2399. doi:10.1093/eurheartj/ehad152
- Aengevaeren VL, Mosterd A, Bakker EA, et al. Exercise Volume Versus Intensity and the Progression of Coronary Atherosclerosis in Middle-Aged and Older Athletes: Findings From the MARC-2 Study. Circulation. 2023;147(13):993-1003. doi:10.1161/CIRCULATIONAHA.122.061173
- Merghani A, Maestrini V, Rosmini S, et al. Prevalence of Subclinical Coronary Artery Disease in Masters Endurance Athletes With a Low Atherosclerotic Risk Profile. Circulation. 2017;136(2):126-137. doi:10.1161/CIRCULATIONAHA.116.026964
- Möhlenkamp S, Lehmann N, Breuckmann F, et al. Running: the risk of coronary events : Prevalence and prognostic relevance of coronary atherosclerosis in marathon runners. Eur Heart J. 2008;29(15):1903-1910. doi:10.1093/eurheartj/ehn163
- Laddu DR, Rana JS, Murillo R, et al. 25-Year Physical Activity Trajectories and Development of Subclinical Coronary Artery Disease as Measured by Coronary Artery Calcium: The Coronary Artery Risk Development in Young Adults (CARDIA) Study. Mayo Clin Proc. 2017;92(11):1660-1670. doi:10.1016/j.mayocp.2017.07.016
- Sung KC, Hong YS, Lee JY, et al. Physical activity and the progression of coronary artery calcification. Heart. 2021;107(21):1710-1716. doi:10.1136/heartjnl-2021-319346
- DeFina LF, Radford NB, Barlow CE, et al. Association of All-Cause and Cardiovascular Mortality With High Levels of Physical Activity and Concurrent Coronary Artery Calcification. JAMA Cardiol. 2019;4(2):174-181. doi:10.1001/jamacardio.2018.4628
- Marijon E, Tafflet M, Antero-Jacquemin J, et al. Mortality of French participants in the Tour de France (1947-2012). Eur Heart J. 2013;34(40):3145-3150. doi:10.1093/eurheartj/eht347
- Sanchis-Gomar F, Olaso-Gonzalez G, Corella D, Gomez-Cabrera MC, Vina J. Increased average longevity among the “Tour de France” cyclists. Int J Sports Med. 2011;32(8):644-647. doi:10.1055/s-0031-1271711
- Ference BA, Kastelein JJP, Ray KK, et al. Association of Triglyceride-Lowering LPL Variants and LDL-C-Lowering LDLR Variants With Risk of Coronary Heart Disease. JAMA. 2019;321(4):364-373. doi:10.1001/jama.2018.20045
- Cholesterol Treatment Trialists’ (CTT) Collaboration, Baigent C, Blackwell L, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376(9753):1670-1681. doi:10.1016/S0140-6736(10)61350-5
- Nissen SE, Lincoff AM, Brennan D, et al. Bempedoic Acid and Cardiovascular Outcomes in Statin-Intolerant Patients. N Engl J Med. 2023;388(15):1353-1364. doi:10.1056/NEJMoa2215024
- Pinkosky SL, Newton RS, Day EA, et al. Liver-specific ATP-citrate lyase inhibition by bempedoic acid decreases LDL-C and attenuates atherosclerosis. Nat Commun. 2016;7:13457. Published 2016 Nov 28. doi:10.1038/ncomms13457
- Cannon CP, Blazing MA, Giugliano RP, et al. Ezetimibe Added to Statin Therapy after Acute Coronary Syndromes. N Engl J Med. 2015;372(25):2387-2397. doi:10.1056/NEJMoa1410489
- Sabatine MS, Giugliano RP, Keech AC, et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N Engl J Med. 2017;376(18):1713-1722. doi:10.1056/NEJMoa1615664
- Schwartz GG, Steg PG, Szarek M, et al. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N Engl J Med. 2018;379(22):2097-2107. doi:10.1056/NEJMoa1801174
- Ray KK, Wright RS, Kallend D, et al. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N Engl J Med. 2020;382(16):1507-1519. doi:10.1056/NEJMoa1912387
- Parker BA, Capizzi JA, Grimaldi AS, et al. Effect of statins on skeletal muscle function. Circulation. 2013;127(1):96-103. doi:10.1161/CIRCULATIONAHA.112.136101
- Di Gioia G, Buzzelli L, Maestrini V, et al. Lipid Profile in Olympic Athletes: Proposal for a “Lipid Athlete Score” as a Clinical Tool to Identify High-Risk Athletes. J Clin Med. 2023;12(23):7449. Published 2023 Nov 30. doi:10.3390/jcm12237449
- D’Ascenzi F, Caselli S, Alvino F, et al. Cardiovascular risk profile in Olympic athletes: an unexpected and underestimated risk scenario. Br J Sports Med. 2019;53(1):37-42. doi:10.1136/bjsports-2018-099530
- Blumenthal RS, Morris PB, Gaudino M, et al. 2026 ACC/AHA/AACVPR/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Dyslipidemia: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J Am Coll Cardiol. 2026;87(19):2624-2757. doi:10.1016/j.jacc.2025.11.016
- Niederseer D, Betschart H, Gähwiler R. Lipid management in athletes – the Swiss approach. SEMS-journal (Sports & Exercise Medicine Switzerland). Published online June 26, 2026. doi:10.34045/SEMS/2026/13.
- Grundy SM, Stone NJ, Bailey AL, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;139(25):e1082-e1143. doi:10.1161/CIR.0000000000000625
- Borén J, Chapman MJ, Krauss RM, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2020;41(24):2313-2330. doi:10.1093/eurheartj/ehz962
- Libby P. The changing landscape of atherosclerosis. Nature. 2021;592(7855):524-533. doi:10.1038/s41586-021-03392-8
- Foote CA, Soares RN, Ramirez-Perez FI, et al. Endothelial Glycocalyx. Compr Physiol. 2022;12(4):3781-3811. Published 2022 Aug 23. doi:10.1002/cphy.c210029
- Li J, Hou B, Tumova S, et al. Piezo1 integration of vascular architecture with physiological force. Nature. 2014;515(7526):279-282. doi:10.1038/nature13701
- Wang L, Luo JY, Li B, et al. Integrin-YAP/TAZ-JNK cascade mediates atheroprotective effect of unidirectional shear flow. Nature. 2016;540(7634):579-582. doi:10.1038/nature20602
- Glagov S, Weisenberg E, Zarins CK, Stankunavicius R, Kolettis GJ. Compensatory enlargement of human atherosclerotic coronary arteries. N Engl J Med. 1987;316(22):1371-1375. doi:10.1056/NEJM198705283162204
- Malek AM, Alper SL, Izumo S. Hemodynamic shear stress and its role in atherosclerosis. JAMA. 1999;282(21):2035-2042. doi:10.1001/jama.282.21.2035
- Davies PF. Flow-mediated endothelial mechanotransduction. Physiol Rev. 1995;75(3):519-560. doi:10.1152/physrev.1995.75.3.519
- Green DJ, Hopman MT, Padilla J, Laughlin MH, Thijssen DH. Vascular Adaptation to Exercise in Humans: Role of Hemodynamic Stimuli. Physiol Rev. 2017;97(2):495-528. doi:10.1152/physrev.00014.2016